
Dantrolen Trockensubstanz 20mg Ampullen 12 Stück buy online
DANTROLEN Trockensub 20 mg
-
2,545.59 CHF
- Price in reward points: 3131

- Availability: Not available
- Product Code: 6811047
- ATC-code M03CA01
- EAN
Ingredients:
Mannitol, Dantrolen natrium 20 mg , Trockensubstanz, Dantrolen, Natronlauge zur pH-Wert-Einstellung.

Description
 Deutsch
Deutsch French
French Italian
ItalianWichtiger Hinweis für die Handhabung:
ZUM AUFZIEHEN BEILIEGENDE EINWEG-FILTRIERVORRICHTUNG VERWENDEN (siehe Rubrik «Sonstige Hinweise»).
Zusammensetzung
Wirkstoffe
Dantrolenum natricum.
Hilfsstoffe
Mannitolum (E421) Natrii hydroxidum (zur pH-Einstellung).
Darreichungsform und Wirkstoffmenge pro Einheit
1 Durchstechflasche Trockensubstanz enthält: 20 mg Dantrolenum natricum, i.v.
Indikationen/Anwendungsmöglichkeiten
Maligne Hyperthermie.
Dosierung/Anwendung
Schnelle i.v. Infusion von 2,5 mg/kg Körpergewicht innerhalb weniger Minuten. Die Infusion solange fortsetzen, wie die klinischen Hauptsymptome Tachykardie, Hypoventilation, anhaltende Hyperazidität und Hyperthermie andauern (Überwachung von pH- und pCO2- erforderlich). Diese Dosis muss bis zu einer Gesamtmenge von 10 mg/kg Körpergewicht in 5-Minuten-Abständen appliziert werden. In den meisten Fällen ist eine Gesamtmenge von 10 mg/kg KG pro 24 Stunden ausreichend. Ist nach einer kumulierten Dosis von 10 mg/kg Körpergewicht ein eindeutiger Effekt nicht zu beobachten, so muss die Diagnose in Zweifel gezogen werden. Gelegentlich kann eine Dosis von über 10 mg/kg Körpergewicht erforderlich sein, um eine maligne Hyperthermie endgültig zu beherrschen. Gesamtdosen von mehr als 40 mg/kg Körpergewicht sind aus Einzelfällen bekannt. Aufgrund dieser Erfahrungen können bei Bedarf in Einzelfällen höhere Dosierungen verabreicht werden.
Kinder und Jugendliche
Gleiche Dosierung bei Kindern und Erwachsenen.
Kontraindikationen
Überempfindlichkeit gegenüber dem Wirkstoff oder einem der sonstige Bestandteile, siehe Abschnitt «Zusammensetzung».
Warnhinweise und Vorsichtsmassnahmen
Nach Gabe von DANTROLEN i.v. kann eine Muskelschwäche mit Beeinträchtigung der Atemfunktion auftreten. Die Atemtätigkeit ist daher zu überwachen.
Der Einsatz von DANTROLEN i.v. bei der Behandlung der malignen Hyperthermie ist kein Ersatz für andere unterstützende Massnahmen. Diese sollen individuell fortgeführt werden.
DANTROLEN i.v. darf nur intravenös infundiert werden. Wegen des hohen pH-Wertes der Lösung (pH 9,5) ist extravasale Injektion unbedingt zu vermeiden, weil sie zu Gewebsnekrosen führen kann. Wegen der Gefahr von Gefässverschlüssen sind intraarterielle Injektionen zu vermeiden. Ausserdem ist aufgrund des möglichen Vorhandenseins von ungelösten Kristallen bzw. Partikeln in der Lösung nach Rekonstitution und der damit verbundenen Gefahr einer Verschlimmerung einer Reaktion an der Infusionsstelle bzw. einer Gewebenekrose, verursacht durch Kristalle in betroffenen Durchstechflaschen, die Verwendung der Filtriervorrichtung beim Aufziehen der Injektionslösung unverzichtbar.
Jede Durchstechflasche DANTROLEN i.v. enthält 3 g Mannitol (zur Einstellung einer isotonischen Lösung). Diese Menge sollte berücksichtigt werden, falls Mannitol zur Vorbeugung und Behandlung von Nierenkomplikationen im Zusammenhang mit maligner Hyperthermie eingesetzt wird.
DANTROLEN i.v. enthält weniger als 1 mmol Natrium (23 mg) pro Durchstechflasche, d.h. es ist nahezu «natriumfrei».
Vorsicht ist beim Auftreten von Hyperkaliämie-Symptomen (muskuläre Paralyse, EKG-Veränderungen, bradykarde Herzrhythmusstörungen) oder bei bereits bestehender Hyperkaliämie (Niereninsuffizienz, Digitalisintoxikation etc.) geboten, da im Tierversuch eine Erhöhung des Serumkaliums durch Dantrolen gezeigt wurde.
Unter DANTROLEN i.v.-Therapie können Leberschädigungen auftreten. Diese sind abhängig von der Dosierung und der Therapiedauer und können einen letalen Verlauf nehmen.
Dosen von mehr als 10 mg Wirkstoff/kg Körpergewicht/24 Stunden können Muskelschwäche, hervorrufen.
Interaktionen
Pharmakodynamische Interaktionen
Tierexperimentelle Untersuchungen weisen auf eine Wechselwirkung von Dantrolen und Verapamil (u.U. auch anderen Calciumantagonisten) in Form von Herzflimmern hin. DANTROLEN i.v. und Verapamil oder andere Calciumantagonisten sollen nicht gleichzeitig angewendet werden.
Wirkung von DANTROLEN i.v. auf andere Arzneimittel
Die gleichzeitige Gabe von DANTROLEN i.v. und nicht-depolarisierenden Muskelrelaxantien wie Vecuronium kann deren Wirkung verstärken.
Schwangerschaft/Stillzeit
Schwangerschaft
Es gibt keine hinreichenden Daten zur Anwendung von Dantrolen bei Schwangeren. In tierexperimentelle Studien fand sich eine Reproduktionstoxizität (siehe «Präklinische Daten»).
Da Erfahrungen mit DANTROLEN i.v.-Behandlung schwangerer Frauen fehlen, sollte eine Anwendung nur erfolgen, wenn dies eindeutig erforderlich ist (vitale Indikation).
Stillzeit
Dantrolen geht in die Plazenta über und wurde in der Muttermilch nachgewiesen. DANTROLEN i.v. sollte bei stillenden Müttern nicht angewendet werden. Wenn eine Behandlung mit DANTROLEN i.v. notwendig ist, sollte abgestillt werden.
Fertilität
Es gibt keine klinischen Daten zu den Auswirkungen von Dantrolen auf die Fertilität. In tierexperimentelle Studien gibt es keinen Nachweis einer direkten Auswirkung von Dantrolen auf die Fertilität (siehe «Präklinische Daten»).
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
DANTROLEN i.v. hat Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen, da es zu Schwindel, Benommenheit und Schwäche führen kann. Dies gilt vor allem im Zusammenwirken mit Alkohol oder anderen das Zentralnervensystem dämpfenden Arzneimitteln (Beruhigungsmittel). Postoperativ sind bis zu 48 Stunden eine Greifschwäche und eine Schwäche der Beinmuskulatur zu erwarten, die sich vor allem beim Treppenlaufen bemerkbar machen kann.
Unerwünschte Wirkungen
Die Häufigkeit folgender Erkrankungen kann auf Grundlage der verfügbaren Daten nicht abgeschätzt werden:
- Erkrankungen des Immunsystems: allergische Reaktionen, Anaphylaxie
- Stoffwechsel- und Ernährungsstörungen: Hyperkaliämie
- Erkrankungen des Nervensystems: Schläfrigkeit, Konvulsionen, Sprachstörung, Schwindel, Schwächegefühl, Kopfschmerzen
- Herzerkrankungen: Herzinsuffizienz, Bradykardie, Tachykardie
- Gefässerkrankungen: Thrombophlebitis
- Erkrankungen der Atemwege, des Brustraums und Mediastinums: Pleuraerguss, Respiratorische Insuffizienz, Atemdepression, Lungenödem
- Erkrankungen des Gastrointestinaltrakts: Übelkeit, Erbrechen, Diarrhö, Bauchschmerzen/-Krämpfe, gastrointestinale Blutungen
- Leber- und Gallenerkrankungen: Ikterus, Hepatitis/Hepatotoxizität, Leberfunktionsstörungen einschliesslich tödlichem Leberversagen, idiosynkratische oder hypertensive Lebererkrankung
- Erkrankungen der Haut und des Unterhautzellgewebes: Hyperhidrose, Urtikaria, Erytheme
- Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen: Muskelschwäche, Muskelermüdung
- Erkrankungen der Nieren und Harnwege: Kristallurie
- Erkrankungen der Geschlechtsorgane und der Brustdrüse: Uterine Hypotonie
- Allgemeine Erkrankungen und Beschwerden am Verabreichungsort: Reaktionen an der Applikationsstelle, Müdigkeit
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Bei der malignen Hyperthermie handelt es sich um eine Notfallsituation, in der die rasche Infusion einer hohen Dosis von DANTROLEN i.v. notwendig ist.
Anzeichen und Symptome
Spezifische Symptome einer DANTROLEN i.v.-Überdosierung sind nicht bekannt. Vorsicht ist bei Zeichen einer Hyperkaliämie geboten.
Behandlung
Der Nutzen einer Dialyse ist nicht bekannt.
Eigenschaften/Wirkungen
ATC-Code
M03CA01
Wirkungsmechanismus
Dantrolen entkoppelt Reiz und Kontraktion des Skelettmuskels durch Hemmung der Calciumfreisetzung aus dem sarkoplasmatischen Retikulum. Es wirkt hier spezifisch, beeinflusst weder die neuro-muskuläre Übertragung noch hat es messbare Wirkung auf die elektrisch erregbare Oberflächenmembran.
Bei dem durch Anästhetika induzierten Syndrom maligne Hyperthermie weisen Anzeichen auf eine genetisch bedingte Anomalie der Muskelzelle hin. Man nimmt an, dass die Triggersubstanzen einen plötzlichen Anstieg des myoplasmatischen Calciums verursachen, indem sie seine Freisetzung verstärken und die Speicherung im sarkoplasmatischen Retikulum verhindern. Der resultierende Anstieg des myoplasmatischen Calcium führt zu einem Hypermetabolismus, der die Ursache der Hyperthermie, der metabolischen Azidose sowie der weiteren Symptome der malignen Hyperthermie ist.
Pharmakodynamik
Dantrolen kann den akuten Katabolismus innerhalb der Muskelzelle verhüten, indem es die Freisetzung von Calcium aus dem sarkoplasmatischen Retikulum in das Myoplasma hemmt. So können die physiologischen, metabolischen und biochemischen Veränderungen, die mit der Krise verbunden sind, umgekehrt oder geschwächt werden.
Klinische Wirksamkeit
Keine Angaben.
Pharmakokinetik
Absorption
Keine Angaben.
Distribution
Maximale Blutkonzentrationen von 4,3 – 6,5 mg/l wurden bei 6 Patienten mit vermuteter maligner Hyperthermie gefunden, denen eine prophylaktische i.v. Infusion von 2,5 mg/kg über eine Zeit von 10 – 30 Min. verabreicht wurde.
Metabolismus
Dantrolen wird in der Leber zu 5-Hydroxydantrolen und Acetylamino-Dantrolen metabolisiert. Der Metabolit 5-Hydroxydantrolen ist pharmakologisch aktiv, hat ca. 50% der Aktivität der Ausgangssubstanz, während Acetylamino-Dantrolen keine muskelrelaxierende Wirkung zeigt.
Elimination
Die Eliminationshalbwertszeit beträgt 12 Std. bei Patienten mit maligner Hyperthermie. Für die diaplazentare Passage wurde ein Faktor von 0,4 gefunden.
Präklinische Daten
Langzeittoxizität
In Studien zur chronischen Toxizität an Ratten, Hunden und Affen führte die orale Gabe von > 30 mg/kg/d Dantrolen (humane Äquivalenzdosis 4,8 mg/kg, 16,2 mg/kg bzw. 9,6 mg/kg) zu Verminderung des Wachstums oder der Körpergewichtsentwicklung und zu toxischen Lebereffekten und möglicherweise Okklusionsnephropathien, die reversibel waren.
Mutagenität
Dantrolen war in in-vitro-Mutagenitätstests (Ames-Tests) bei einigen Bakterienstämmen in An- und Abwesenheit von Rattenleberhomogenat positiv.
Karzinogenität
Diätetische Dosen von Dantrolen bei Ratten in Dosen von bis zu 60 mg/kg/Tag (humane Äquivalenzdosis 9,6 mg/kg) über einen Zeitraum von bis zu 18 Monaten führten zu einer Zunahme gutartiger hepatischer Lymphgefässneubildungen, zu vermehrten hepatischen Lymphangiomen und hepatischen Angiosarkomen und nur bei weiblichen Tieren zu einer Zunahme von Brusttumoren.
Die Relevanz dieser Daten für die klinische Anwendung von Dantrolen ist nicht bekannt.
Reproduktionstoxizität
Bei männlichen und weiblichen erwachsenen Ratten hatte Dantrolen bis zu einer oralen Dosis von 45 mg/Körpergewicht/Tag (humane Äquivalenzdosis 7,3 mg/kg/Tag) keine nachteiligen Auswirkungen auf die Fertilität oder die allgemeine Fortpflanzungsfähigkeit. Die Verabreichung von Dantrolen an trächtige Ratten (ab 20 mg/kg/Tag; humane Äquivalenzdosis 3,2 mg/kg/Tag) und Kaninchen (45 mg/kg/Tag; humane Äquivalenzdosis 14,5 mg/kg/Tag) führte zu einer verstärkten Bildung ein- oder beidseitiger überzähliger Rippen bei den Jungtieren.
Sonstige Hinweise
Die zubereitete DANTROLEN i.v.-Lösung darf nicht mit anderen Infusionslösungen gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «verwendbar bis» bezeichneten Datum verwendet werden.
Nach Rekonstitution und Filtration ist die Lösung gebrauchsfertig und muss SOFORT verwendet werden. Übriggebliebene Lösung entsorgen.
Besondere Lagerungshinweise
Für Kinder unzugänglich aufbewahren.
Trockensubstanz nicht über 25 °C lagern.
Die Lösung ist vor direktem Licht zu schützen und zwischen 15-25 °C zu lagern.
Hinweise für die Handhabung
Lösungsvorschrift: Zu jeder Durchstechflasche DANTROLEN i.v. werden 60 ml Wasser für Injektionszwecke gegeben und solange geschüttelt, bis die Lösung klar ist.
Wichtig - vor Gebrauch lesen:
Rekonstitution von DANTROLEN i.v. (Dantrolen-Natrium)
Die rekonstituierte DANTROLEN -i.v.-Lösung muss vor der Anwendung am Patienten filtriert werden, um Partikelfreiheit zu gewährleisten. Dazu ist wie folgt vorzugehen:
- Die Durchstechflasche mit 60 ml Wasser für Injektionszwecke rekonstituieren. Zur Zugabe des Wassers NICHT die Einweg-Filtriervorrichtung verwenden.
- Nach der Rekonstitution muss die Lösung beim Aufziehen in die Spritze durch die beiliegende Filtriervorrichtung gefiltert werden. Bei jeder neuen DANTROLEN i.v. Durchstechflasche eine NEUE Einweg-Filtriervorrichtung verwenden. Die Lösung muss innerhalb 6 Stunden verwendet werden, aber unmittelbar vor der Verwendung filtriert werden.
- Die Einweg-Filtriervorrichtung von der Spritze entfernen, durch eine i.v. Kanüle oder ein Verabreichungsset ersetzen und das Präparat SOFORT nach Filtration verabreichen.
- Die Filtriervorrichtung und die Durchstechflasche mit dem Produkt in einem geprüften Sicherheitsbehälter (Sharps Collector) entsorgen.
Achtung: Nur für i.v. Infusion. Nur die beiliegende Einweg-Filtriervorrichtung verwenden. Mehrfache Verwendung der Filtriervorrichtung kann zu Infektionen oder anderen Erkrankungen/Verletzungen führen. Die Filtriervorrichtung nicht autoklavieren. Bei Beschädigung von Einzelverpackungen nicht verwenden.
Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Information über Rekonstitution und Filtration von DANTROLEN i.v.
| 1) Eine Kanüle auf eine Spritze setzen und 60 ml Wasser für Injektionszwecke aufziehen. |
| 2) Eine DANTROLEN i.v. Durchstechflasche mit aufgezogenem Wasser für Injektionszwecke rekonstituieren. Leicht schütteln bis das Pulver vollständig gelöst ist. Die Kanüle verwerfen. |
| 3) Die Sicherheitskappe entfernen und die Spitze der Einwegfiltriervorrichtung in die Durchstechflasche stecken. |
| 4) Die Spritze anschliessen und die gesamte 60 ml rekonstituierte Lösung aus der Durchstechflasche in die Spritze ziehen und im Anschluss die Filtriervorrichtung verwerfen. |
| 5) Die Spritze mit der filtrierten rekonstituierten Lösung direkt mit der intravenösen Kanüle oder einem Verabreichungsset verbinden. Abhängig von der klinischen Notwendigkeit wird das Produkt entweder sofort verabreicht oder als manuelle Infusion bzw. über eine Pumpe. Siehe Abschnitt Dosierung/Anwendung für die maximale Dosis. |




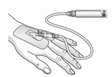
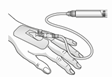
Warnhinweis für die Handhabung:
| Achtung: Für den Transfer der filtrierten Lösung von der Spritze in die intravenöse Kanüle bzw. Verabreichungsset nicht die Filtriervorrichtung verwenden! |
Zulassungsnummer
45217 (Swissmedic)
Zulassungsinhaberin
Norgine AG, 6005 Luzern
Stand der Information
Februar 2021
Indicazioni importanti per la manipolazione:
PER L'ASPIRAZIONE UTILIZZARE IL DISPOSITIVO DI FILTRAZIONE MONOUSO IN DOTAZIONE (vedere inoltre la rubrica «Altre indicazioni»).
Composizione
Principi attivi
Dantrolenum natricum.
Sostanze ausiliarie
Mannitolum (E421) Natrii hydroxidum (per la regolazione del pH).
Forma farmaceutica e quantità di principio attivo per unità
1 flaconcino di sostanza secca contiene: 20 mg di Dantrolenum natricum, e.v.
Indicazioni/Possibilità d'impiego
Ipertermia maligna.
Posologia/Impiego
Infusione rapida e.v. di 2,5 mg/kg di peso corporeo nell'arco di pochi minuti. Proseguire l'infusione fino a quando i principali sintomi clinici di tachicardia, ipoventilazione, iperacidità persistente e ipertermia persistono (occorre monitorare i valori di pH e pCO2). Ripetere la somministrazione a intervalli di 5 minuti fino a raggiungere una quantità complessiva di 10 mg/kg di peso corporeo. Nella maggior parte dei casi è sufficiente una quantità complessiva di 10 mg/kg di peso corporeo per 24 ore. Se una volta raggiunta la dose cumulativa di 10 mg/kg di peso corporeo non si osserva un netto miglioramento, si deve rivalutare la diagnosi. Talvolta può essere necessario somministrare una dose superiore a 10 mg/kg di peso corporeo al fine di controllare definitivamente l'ipertermia maligna. Dosi complessive superiori a 40 mg/kg di peso corporeo sono note da casi isolati. Sulla base di tali dati, è possibile somministrare dosaggi più elevati in casi isolati secondo necessità.
Bambini e adolescenti
Posologia identica per bambini e adulti.
Controindicazioni
Ipersensibilità al principio attivo o a una delle sostanze ausiliarie, vedere la sezione «Composizione».
Avvertenze e misure precauzionali
Dopo la somministrazione di DANTROLEN i.v., può comparire una debolezza muscolare associata a compromissione della funzione respiratoria. Pertanto, occorre monitorare l'attività respiratoria. L'uso di DANTROLEN i.v. nel trattamento dell'ipertermia maligna non sostituisce altre misure di supporto. Tali misure devono essere proseguite individualmente.
DANTROLEN i.v. deve essere somministrato solo per infusione endovenosa (e.v.). Essendo la soluzione molto acida (pH 9,5), occorre fare molta attenzione a evitare l'iniezione intrarteriosa, perché potrebbe causare necrosi tissutale. A causa del pericolo di occlusione vascolare, la somministrazione intra-arteriosa deve essere evitata. Inoltre, dopo la ricostituzione, alcuni flaconcini potrebbero contenere particelle o cristalli insoluti, con conseguente aumento del rischio di reazioni nel sito di iniezione o di necrosi tissutale. Per tale motivo, è indispensabile prelevare la soluzione iniettabile facendo uso del dispositivo di filtrazione.
Ciascun flaconcino di DANTROLEN i.v. contiene 3 g di mannitolo (per ottenere una soluzione isotonica). Questa quantità deve essere considerata qualora il mannitolo sia impiegato per prevenire e trattare le complicazioni renali associate all'ipertermia maligna.
DANTROLEN i.v. contiene meno di 1 mmol di sodio (23 mg) per flaconcino, cioèessenzialmente «senzia sodio».
Occorre prestare cautela in presenza di sintomi di iperkaliemia (paralisi muscolare, alterazioni all'ECG, aritmie bradicardiche) oppure di iperkaliemia preesistente (insufficienza renale, intossicazione digitalica ecc.), giacché negli esperimenti sugli animali è stato rilevato un innalzamento del potassio sierico a seguito della terapia con dantrolene.
La terapia con DANTROLEN i.v. può causare danni epatici, la cui entità (che nei casi più gravi può avere esiti letali) dipende dal dosaggio e dalla durata della terapia.
Dosi di principio attivo superiori a 10 mg/kg di peso corporeo/24 ore possono causare debolezza muscolare.
Interazioni
Interazioni farmacodinamiche
Gli studi sperimentali condotti su animali dimostrano l'esistenza di un'interazione tra dantrolene e verapamil (forse anche con altri calcio-antagonisti), che si manifesta sotto forma di fibrillazione. DANTROLEN i.v. e verapamil o altri calcio-antagonisti non devono essere impiegati per terapie concomitanti.
Effetto di DANTROLEN i.v. su altri medicamenti
La somministrazione concomitante di DANTROLEN i.v. e di miorilassanti non depolarizzanti quali vecuronio può amplificarne l'effetto.
Gravidanza/Allattamento
Gravidanza
Non sono disponibili dati sufficienti sull'utilizzo di dantrolene nelle donne in gravidanza. Negli studi sperimentali condotti sugli animali è stata riscontrata tossicità per la riproduzione (vedere «Dati preclinici»).
Poiché non sono ancora disponibili dati relativi alla somministrazione di DANTROLEN i.v. in donne in gravidanza, se ne consiglia l'uso solamente nel caso in cui sia strettamente necessario (indicazione vitale).
Allattamento
Dantrolene attraversa la barriera placentare e la sua presenza è stata rilevata nel latte materno. Non somministrare DANTROLEN i.v. alle donne che allattano. Se il trattamento con DANTROLEN i.v. è indispensabile, sospendere l'allattamento.
Fertilità
Non sono disponibili dati clinici sugli effetti di dantrolene sulla fertilità. Negli studi sperimentali condotti sugli animali non sono state riscontrate prove di un effetto diretto di dantrolene sulla fertilità (vedere «Dati preclinici»).
Effetti sulla capacità di condurre veicoli e sull'impiego di macchine
DANTROLEN i.v. altera la capacità di condurre veicoli o di impiegare macchine, giacché può indurre vertigine, stordimento e debolezza. Ciò si verifica, soprattutto, in associazione con alcol o altri medicamenti che inibiscono il sistema nervoso centrale (sedativi). Dopo l'intervento possono comparire e persistere fino a 48 ore una riduzione della forza prensile delle mani e una debolezza muscolare agli arti inferiori, che si rende evidente soprattutto quando si salgono le scale.
Effetti indesiderati
La frequenza delle seguenti patologie non può essere definita sulla base dei dati disponibili:
- Disturbi del sistema immunitario: reazioni allergiche, anafilassi
- Disturbi del metabolismo e della nutrizione: iperkaliemia
- Patologie del sistema nervoso: sonnolenza, convulsioni, disturbi del linguaggio, vertigine, sensazione di debolezza, cefalea
- Patologie cardiache: insufficienza cardiaca, bradicardia, tachicardia
- Patologie vascolari: tromboflebite
- Patologie respiratorie, toraciche e mediastiniche: versamento pleurico, insufficienza respiratoria, depressione respiratoria, edema polmonare
- Patologie gastrointestinali: nausea, vomito, diarrea, dolori/crampi addominali, emorragia gastrointestinale
- Patologie epatobiliari: ittero, epatite/epatotossicità, compromissione epatica compresa insufficienza epatica fatale, malattia epatica idiosincratica o ipertensiva
- Patologie della cute e del tessuto sottocutaneo: iperidrosi, orticaria, eritema
- Patologie del sistema muscoloscheletrico e del tessuto connettivo: debolezza muscolare, affaticamento muscolare
- Patologie renali e urinarie: cristalluria
- Patologie dell'apparato riproduttivo e della mammella: ipotensione uterina
- Patologie generali e condizioni relative alla sede di somministrazione: reazioni nella sede di applicazione, stanchezza
La notifica di effetti collaterali sospetti dopo l'omologazione del medicamento è molto importante. Consente una sorveglianza continua del rapporto rischio-beneficio del medicamento. Chi esercita una professione sanitaria è invitato a segnalare qualsiasi effetto indesiderato sospetto, nuovo o serio, attraverso il portale online ElViS (Electronic Vigilance System). Maggiori informazioni sul sito www.swissmedic.ch.
Posologia eccessiva
L'ipertermia maligna è un'emergenza medica che richiede l'infusione rapida di una dose elevata di DANTROLEN i.v.
Segni e sintomi
Non si conoscono sintomi specifici di un sovradosaggio da DANTROLEN i.v. Occorre prestare cautela in presenza di segni d'una iperkaliemia.
Trattamento
Non è noto il vantaggio di sottoporre il paziente a dialisi.
Proprietà/Effetti
Codice ATC
M03CA01
Meccanismo d'azione
Dantrolene inibisce il rilascio del calcio dal reticolo sarcoplasmatico, riducendo in tal modo l'accoppiamento eccitazione-contrazione nel muscolo scheletrico. In tal caso, agisce in maniera specifica, senza influire sulla trasmissione neuromuscolare e senza avere un effetto misurabile sulla membrana superficiale elettricamente eccitabile.
Nell'ipertermia maligna come sindrome indotta da anestetici, si evidenziano segni indicativi di un'anomalia di origine genetica della cellula muscolare. Si presume che le sostanze scatenanti inducano un maggiore rilascio del calcio mioplasmatico conseguente ad un improvviso aumento della sua concentrazione, inibendone in tal modo l'accumulo nel reticolo sarcoplasmatico. Il conseguente aumento del livello del calcio mioplasmatico induce un ipermetabolismo che è causa dell'ipertermia, dell'acidosi metabolica e degli altri sintomi dell'ipertermia maligna.
Farmacodinamica
Dantrolene è in grado di prevenire il catabolismo acuto all'interno della cellula muscolare, inibendo il rilascio del calcio dal reticolo sarcoplasmatico nel mioplasma. In questo modo è possibile invertire o ridurre le modificazioni fisiologiche, metaboliche e biochimiche legate alla crisi.
Efficacia clinica
Non sono disponibili dati in merito.
Farmacocinetica
Assorbimento
Non sono disponibili dati in merito.
Distribuzione
La massima concentrazione ematica di 4,3–6,5 mg/l è stata rilevata in 6 pazienti con sospetta ipertermia maligna, che erano stati sottoposti a infusione profilattica e.v. L'infusione di 2,5 mg/kg è stata somministrata per un periodo di 10–30 min.
Metabolismo
Dantrolene viene metabolizzato nel fegato in 5-idrossidantrolene e acetilammiino-dantrolene. Il metabolita 5-idrossidantrolene è farmacologicamente attivo, ha circa il 50% dell'attività della sostanza originale, mentre l'acetilammino-dantrolene non mostra alcun effetto miorilassante.
Eliminazione
I pazienti affetti da ipertermia maligna presentano un'emivita di eliminazione pari a 12 ore. Per il passaggio diaplacentare è stato rilevato un fattore di 0,4.
Dati preclinici
Tossicità a lungo termine
In studi di tossicità cronica su ratti, cani e scimmie, la somministrazione orale di > 30 mg/kg/die di dantrolene (dose umana equivalente 4,8 mg/kg, 16,2 mg/kg e 9,6 mg/kg, rispettivamente) ha provocato una diminuzione della crescita o dello sviluppo del peso corporeo ed effetti epatotossici, oltre a possibili nefropatie ostruttive, che si sono dimostrate reversibili.
Mutagenicità
Dantrolene è risultato positivo ai test di mutagenicità in vitro (test di Ames) su alcuni ceppi batterici in presenza/assenza di omogenato epatico di ratto.
Cancerogenicità
Dosi dietetiche di dantrolene sodico somministrate in ratti a dosi fino a 60 mg/kg/die (dose umana equivalente 9,6 mg/kg) per un massimo di 18 mesi hanno provocato un aumento della neoformazione di vasi linfatici epatici benigni, un aumento dei linfangiomi epatici e degli angiosarcomi epatici, oltre a un aumento dei tumori mammari solo nelle femmine.
La rilevanza di questi dati per l'uso clinico di dantrolene non è nota.
Tossicità per la riproduzione
Nei ratti adulti maschi e femmine, dantrolene non ha avuto effetti negativi sulla fertilità o sulla capacità riproduttiva generale fino a una dose orale di 45 mg/peso corporeo/die (dose umana equivalente 7,3 mg/kg/die). La somministrazione di dantrolene in femmine gravide di ratto (a partire da 20 mg/kg/die; dose umana equivalente 3,2 mg/kg/die) e di coniglio (45 mg/kg/die; dose umana equivalente 14,5 mg/kg/die) ha portato ad un aumento della formazione di costole soprannumerarie unilaterali o bilaterali negli animali giovani.
Altre indicazioni
Non mescolare la soluzione di DANTROLEN i.v. preparata con altre soluzioni per infusione.
Stabilità
Il medicamento non deve essere utilizzato oltre la data indicata con «EXP» sulla confezione.
Una volta ricostituita e filtrata, la soluzione è pronta per l'uso e deve essere somministrata IMMEDIATAMENTE. Scartare la soluzione eccedente.
Precauzioni particolari per la conservazione
Tenere fuori dalla portata dei bambini.
Non conservare la sostanza secca a temperature superiori a 25°C.
Proteggere la soluzione dalla luce diretta e conservarla alla temperatura di 15–25°C.
Indicazioni per la manipolazione
Istruzioni per la miscelazione: aggiungere a ciascun flaconcino di DANTROLEN i.v. 60 ml di acqua per soluzione iniettabile e agitare fino a che la soluzione appare chiara.
Importante - leggere prima dell'uso:
Ricostituzione di DANTROLEN i.v. (dantrolene sodico)
Filtrare la soluzione di DANTROLEN i.v. ricostituita prima di somministrarla al paziente, per garantire che sia libera da particelle. Per fare ciò, procedere come segue:
- Ricostituire il flaconcino con 60 ml di acqua per soluzione iniettabile. A questo scopo NON utilizzare il dispositivo di filtrazione monouso.
- Dopo la ricostituzione, filtrare la soluzione aspirandola nella siringa mediante il dispositivo di filtrazione fornito in dotazione. Per ciascun nuovo flaconcino di DANTROLEN i.v. utilizzare un dispositivo di filtrazione monouso NUOVO. Utilizzare la soluzione entro 6 ore, ma filtrarla solo immediatamente prima dell'uso.
- Rimuovere il dispositivo di filtrazione monouso dalla siringa, sostituirlo con una cannula e.v. o un set per somministrazione e somministrare il preparato IMMEDIATAMENTE dopo la filtrazione.
- Smaltire il dispositivo di filtrazione e il flaconcino contenente il prodotto in un contenitore di sicurezza certificato (Sharps Collector).
Attenzione: solo per infusione e.v. Utilizzare solo il dispositivo di filtrazione fornito in dotazione. L'uso ripetuto del dispositivo di filtrazione può causare infezioni o altre malattie/lesioni. Non riporre in autoclave il dispositivo di filtrazione. Non utilizzare le confezioni singole se danneggiate.
Smaltire il medicamento non utilizzato o il materiale di scarto conformemente alle normative nazionali in materia.
Informazioni sulla ricostituzione e sulla filtrazione di DANTROLEN i.v.
| 1) Applicare una cannula sulla siringa e aspirare 60 ml di acqua per soluzione iniettabile. |
| 2) Ricostituire un flaconcino di DANTROLEN i.v. aspirando acqua per soluzione iniettabile. Agitare leggermente fino a che la polvere non si dissolve completamente. Eliminare la cannula. |
| 3) Rimuovere il tappo di sicurezza e introdurre la punta del dispositivo di filtrazione monouso nel flaconcino. |
| 4) Avvitare la siringa, aspirare tutti i 60 ml di soluzione ricostituita dal flaconcino, quindi smaltire il dispositivo di filtrazione monouso. |
| 5) Collegare la siringa contenente la soluzione ricostituita filtrata direttamente alla cannula per endovena o ad un set per somministrazione. A seconda della situazione clinica, somministrare il prodotto direttamente oppure sotto forma di infusione manuale o ancora mediante una pompa. Vedere la sezione Posologia/impiego per la dose massima. |




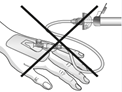
Avvertenze per la manipolazione:
| Attenzione: Non utilizzare il dispositivo di filtrazione per trasferire la soluzione filtrata dalla siringa alla cannula per endovena o al set per somministrazione! |
Numero dell'omologazione
45217 (Swissmedic)
Titolare dell’omologazione
Norgine AG, 6005 Lucerna
Stato dell'informazione
Febbraio 2021
Consignes importantes de manipulation:
POUR LE PRÉLÈVEMENT, UTILISER LE DISPOSITIF DE FILTRATION À USAGE UNIQUE FOURNI (voir la rubrique «Remarques particulières»).
Composition
Principes actifs
Dantrolenum natricum.
Excipients
Mannitolum (E421), Natrii hydroxidum (pour ajustement du pH).
Forme pharmaceutique et quantité de principe actif par unité
1 flacon perforable de substance sèche contient: 20 mg Dantrolenum natricum i.v.
Indications/Possibilités d’emploi
Hyperthermie maligne.
Posologie/Mode d’emploi
Perfusion i.v. rapide de 2,5 mg/kg de poids corporel en l'espace de quelques minutes. Poursuivre la perfusion tant que les principaux signes cliniques de tachycardie, hypoventilation, hyperacidité persistante et hyperthermie persistent (surveillance nécessaire du pH et de la pCO2). Cette posologie doit être répétée avec 5 minutes d'intervalle jusqu'à une dose totale de 10 mg/kg de poids corporel. Dans la plupart des cas, une quantité totale de 10 mg/kg du poids corporel (pc) par jour (24 heures) suffit. Lorsqu'une dose cumulée de 10 mg/kg de poids corporel ne produit pas l'effet attendu, le diagnostic doit être remis en cause. Une dose cumulée de plus de 10 mg/kg de poids corporel peut parfois être nécessaire pour maîtriser définitivement une hyperthermie maligne. Des doses totales de plus de 40 mg/kg de poids corporel sont connues dans des cas isolés. Sur la base de cette expérience, des doses plus élevées peuvent être administrées dans des cas individuels si nécessaire.
Enfants et adolescents
La posologie est identique pour enfants et adultes.
Contre-indications
Hypersensibilité au principe actif ou à l'un des autres composants, voir au chapitre «Composition».
Mises en garde et précautions
L'administration de DANTROLEN i.v. peut entraîner une faiblesse musculaire entravant la fonction respiratoire. La respiration doit donc être contrôlée.
L'utilisation de DANTROLEN i.v. dans le traitement de l'hyperthermie maligne ne remplace pas les autres mesures de soutien. Ceux-ci doivent être poursuivis individuellement.
DANTROLEN i.v. ne peut être administré que par voie intraveineuse. Le pH élevé de la solution – 9,5 – interdit l'injection paraveineuse qui pourrait provoquer une nécrose des tissus. La perfusion intra-artérielle est à éviter en raison du danger d'oblitération vasculaire.
En outre, l'utilisation d'un dispositif de filtration pour le prélèvement de la solution injectable est indispensable en raison de la présence éventuelle de cristaux non dissous ou de particules dans la solution après reconstitution et du risque qui en résulte d'une aggravation de la réaction au site de perfusion, resp. d'une nécrose des tissus provoquée par la présence de cristaux dans les flacons concernés.
Chaque flacon perforable de DANTROLEN i.v. contient 3 g de mannitol (pour l'ajustage d'une solution isotonique). Cette quantité doit être prise en compte si le mannitol est utilisé pour prévenir et traiter les complications rénales associées à l'hyperthermie maligne.
Le dantrolène i.v. contient moins de 1 mmol de sodium (23 mg) par flacon, c'est-à-dire qu'il est pratiquement «sans sodium».
La prudence s'impose en cas d'apparition de symptômes d'hyperkaliémie (paralysie musculaire, modification de l'ECG, arythmies cardiaques avec bradycardie) ou en cas d'hyperkaliémie pré-existante (insuffisance rénale, intoxication digitalique, etc.); car les essais sur les animaux ont révélé que le dantrolène entraînait une augmentation du potassium sérique.
Des doses supérieures à 10 mg de principe actif par kg de poids corporel et par 24 h peuvent entraîner une faiblesse musculaire.
Interactions
Interactions pharmacodynamiques
L'expérimentation animale suggère la possibilité d'une interaction entre dantrolène et le vérapamil (éventuellement aussi avec d'autres antagonistes du calcium) provoquant une fibrillation. DANTROLEN i.v. ainsi que le vérapamil ou d'autres antagonistes du calcium ne doivent donc pas être administrés en même temps.
Effet de DANTROLEN iv sur d'autres médicaments
La prise concomitante de DANTROLEN i.v. et d'un relaxant musculaire non-dépolarisant tel que le vécuronium peut renforcer leur effet respectif.
Grossesse/Allaitement
Grossesse
Les données sur l'utilisation du dantrolène chez la femme enceinte sont insuffisantes. Des études sur les animaux ont révélé une toxicité pour la reproduction (voir «Données précliniques»).
Il n'existe pas de données disponibles sur le traitement avec DANTROLEN i.v. chez la femme enceinte, c'est pourquoi l'administration ne devrait uniquement être effectuée que si elle s'avère absolument nécessaire (indication vitale).
Allaitement
Dantrolène traverse le placenta et a pu être décelé dans le lait maternel. DANTROLEN i.v. ne devrait pas être administré aux femmes qui allaitent. Si un traitement avec DANTROLEN i.v. est nécessaire, l'allaitement devrait être interrompu.
Fertilité
Il n'existe pas de données cliniques concernant les effets du dantrolène sur la fertilité. Les études sur les animaux ne fournissent pas de preuves d'un effet direct du dantrolène sur la fertilité (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machines
DANTROLEN i.v. affecte l'aptitude à conduire et à utiliser des machines, en raison de risques de vertige, obnubilation et faiblesse. Cela est particulièrement vrai en cas d'association avec l'alcool ou d'autres médicaments qui dépriment le système nerveux central (sédatifs). Jusqu'à 48 heures post opératoire, il faut s'attendre à une faiblesse de préhension et une faiblesse de la musculature des jambes, qui se manifeste principalement en montant les escaliers.
Effets indésirables
Comme il n'existe pas de données à ce sujet, la fréquence des troubles suivants ne peut pas faire l'objet d'une estimation:
- Affections du système immunitaire: réactions allergiques, anaphylaxie
- Troubles du métabolisme et de la nutrition: hyperkaliémie
- Affections du système nerveux: somnolence, convulsions, troubles de l'élocution, vertiges, sensation de faiblesse, maux de tête
- Affections cardiaques: insuffisance cardiaque, bradycardie, tachycardie
- Affections vasculaires: thrombophlébite
- Affections respiratoires, thoraciques et médiastinales: épanchement pleural, insuffisance respiratoire, dépression respiratoire, œdème pulmonaire
- Affections gastro-intestinaux: nausées, vomissements, diarrhées, douleurs/crampes abdominales, hémorragies gastro-intestinales
- Affections hépatobiliaries: ictère, hépatite/hépatotoxicité, dysfonctionnement hépatique y compris insuffisance hépatique fatale, maladie hépatique idiosyncrasique ou hypertensive
- Affections de la peau et du tissu sous-cutané: hyperhidrose, urticaire, érythème
- Affections musculosquelettiques et du tissu conjonctif: faiblesse musculaire, fatigue musculaire
- Affections du rein et des voies urinaires: cristallurie
- Affections des organes de reproduction et du sein: hypotonie utérine
- Troubles généraux et anomalies au site d'administration: réactions au point d'administration, fatigue
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
L'hyperthermie maligne est une situation d'urgence, dans laquelle une infusion rapide d'une dose élevée de DANTROLEN i.v. est nécessaire.
Signes et symptômes
Des symptômes spécifiques liés à un surdosage de DANTROLEN i.v. ne sont pas connus. Il faut rester vigilant aux signes d'une hyperkaliémie.
Traitement
L'utilisation d'une dialyse n'est pas connue.
Propriétés/Effets
Code ATC
M03CA01
Mécanisme d'action
Le dantrolène dissocie le couplage excitation-contraction du muscle strié en inhibant la libération du calcium stocké dans les membranes du réticulum sarcoplasmique. Il exerce à cet égard une action ciblée et n'influence pas la transmission neuromusculaire. Il n'exerce également pas d'effet mesurable sur l'excitabilité de la membrane sarcoplasmique.
Le syndrome d'hyperthermie maligne, qui se manifeste en réponse à l'inhalation d'agents anesthésiques, renvoie à une anomalie génétique du transport calcique dans la cellule musculaire. On estime que les substances déclenchantes provoquent une augmentation subite du taux de calcium myoplasmique en renforçant sa libération et inhibant son stockage dans le réticulum sarcoplasmique. Cette augmentation du taux de calcium myoplasmique aboutit à une activité métabolique intense qui est à l'origine de l'hyperthermie, de l'acidose métabolique et des autres symptômes caractérisant l'hyperthermie maligne.
Pharmacodynamique
Le dantrolène peut prévenir les processus cataboliques aigus au sein de la cellule musculaire en inhibant la libération du calcium stocké dans les membranes du réticulum sarcoplasmique. Les altérations physiologiques, métaboliques et biochimiques accompagnant l'hyperthermie maligne peuvent ainsi être inversées ou atténuées.
Efficacité clinique
Aucune information.
Pharmacocinétique
Absorption
Aucune information.
Distribution
Des concentrations plasmatiques de l'ordre de 4,3–6,5 mg/l ont été trouvées chez 6 patients à hyperthermie maligne ayant reçu à titre préventif une perfusion i.v. de 2,5 mg/kg pendant 10 à 30 min.
Métabolisme
Le dantrolène est métabolisé dans le foie en 5-hydroxydantrolène et acétylamino-dantrolène. L'activité du 5-hydroxydantrolène représente environ 50% de celle de la molécule mère tandis que l'acétylamino-dantrolène n'a pas d'activité myorelaxante.
Élimination
La demi-vie biologique du dantrolène est de 12 h chez les patients atteints d'hyperthermie maligne. Un facteur de 0,4 a été trouvé pour le passage de la barrière placentaire.
Données précliniques
Toxicité à long terme
Lors des essais de toxicité chronique chez le rat, le chien et le singe, l'administration orale de > 30 mg/kg/d de dantrolène (doses équivalentes pour l'homme respectivement de 4,8 mg/kg, 16,2 mg/kg et 9,6 mg/kg) a entraîné une diminution de la croissance ou de la prise de poids, ainsi que des effets toxiques sur le foie et de probables néphropathies occlusives réversibles.
Mutagénicité
Le dantrolène s'est révélé positif dans les essais de mutagénicité in vitro (tests d'Ames) sur certaines souches bactériennes en présence et en l'absence d'homogénat de foie de rat.
Cancérogénicité
L'administration de dantrolène par voie orale chez le rat, à des doses allant jusqu'à 60 mg/kg/jour (dose équivalente pour l'homme de 9,6 mg/kg) pendant une période allant jusqu'à 18 mois a entraîné une augmentation des régénérations hépatiques bénignes des vaisseaux lymphatiques, une augmentation des lymphangiomes hépatiques et des angiosarcomes hépatiques, et une augmentation des tumeurs mammaires chez les animaux femelles uniquement.
La pertinence de ces données pour l'utilisation clinique du dantrolène n'est pas connue.
Toxicité pour la reproduction
Chez les rats adultes mâles et femelles, le dantrolène n'a pas eu d'effets indésirables sur la fertilité ou la capacité générale de reproduction jusqu'à une dose orale de 45 mg/pc/jour (dose équivalente pour l'homme de 7,3 mg/kg/jour). L'administration de dantrolène à des rats femelles en gestation (à partir de 20 mg/kg/jour; dose équivalente chez l'homme de 3,2 mg/kg/jour) et à des lapines (45 mg/kg/jour; dose équivalente chez l'homme de 14,5 mg/kg/jour) a entraîné une augmentation de la formation de côtes surnuméraires unilatérales ou bilatérales chez les jeunes.
Remarques particulières
Une fois préparée, la solution de DANTROLEN i.v. ne doit pas être mélangée à d'autres solutions de perfusion.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date de péremption imprimée sur l'emballage avec la mention «Verwendbar bis».
Après reconstitution et filtration, la solution est prête à l'emploi et doit être utilisée IMMÉDIATEMENT. Éliminer la solution restante.
Remarques particulières concernant le stockage
Tenir hors de portée des enfants.
Conserver la substance sèche au-dessous de 25 °C.
Protéger la solution de la lumière directe et la conserver entre 15-25 °C.
Remarques concernant la manipulation
Instructions pour la préparation de la solution: Ajouter 60 ml d'eau pour préparations injectables à chaque flacon de DANTROLEN i.v. et agiter jusqu'à ce que la solution soit claire.
Informations importantes à lire avant l'emploi:
Reconstitution de DANTROLEN i.v. (dantrolène-sodique)
La solution de DANTROLEN i.v. reconstituée doit être filtrée avant d'être administrée au patient afin de garantir l'absence de particules. Pour ce faire, procédez comme suit:
- Reconstituez le flacon avec 60 ml d'eau pour préparations injectables. Pour ajouter l'eau, NE PAS utiliser le dispositif de filtration à usage unique.
- Après la reconstitution, la solution doit être filtrée au moyen du dispositif de filtration fourni lors du prélèvement dans la seringue. Lorsqu'on utilise un nouveau flacon de DANTROLEN i.v., utiliser systématiquement un NOUVEAU dispositif de filtration à usage unique. La solution doit être utilisée dans les 6 heures suivant la reconstitution, mais doit être filtrée juste avant utilisation.
- Enlevez le dispositif de filtration à usage unique de la seringue avant de connecter celle-ci à une canule intraveineuse ou un set d'administration et administrer la préparation IMMÉDIATEMENT après filtration.
- Jetez le dispositif de filtration et le flacon avec le produit dans un collecteur pour objets pointus et tranchants approuvé.
Attention: Pour injection par voie intraveineuse uniquement. Utiliser uniquement le dispositif de filtration à usage unique fourni. Une réutilisation du dispositif de filtration peut engendrer une infection ou une autre maladie/blessure. Ne pas stériliser le dispositif de filtration en autoclave. Ne pas utiliser si l'emballage individuel est endommagé.
Le médicament non utilisé et les déchets doivent être éliminés conformément aux dispositions nationales en vigueur.
Informations sur la reconstitution et la filtration de DANTROLEN i.v.
| 1) Mettre une canule sur une seringue et prélever 60 ml d'eau pour préparations injectables. |
| 2) Reconstituer un flacon de DANTROLEN i.v. avec l'eau pour préparations injectables prélevée auparavant. Agiter légèrement jusqu'à ce que la poudre soit entièrement dissoute. Jeter la canule. |
| 3) Retirer la capsule de sécurité et insérer la pointe du dispositif de filtration à usage unique dans le flacon. |
| 4) Connecter la seringue et aspirer la totalité des 60 ml de solution reconstituée du flacon dans la seringue, puis jeter le dispositif de filtration. |
| 5) Connecter la seringue contenant la solution reconstituée filtrée directement à la canule intraveineuse ou à un set d'administration. En fonction de la nécessité clinique, le produit est administré immédiatement ou par perfusion manuelle, resp. au moyen d'une pompe. Voir la rubrique Posologie/Mode d'emploi pour la dose maximale. |




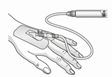
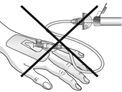
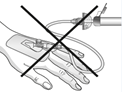
Mises en garde concernant la manipulation:
| Attention: Ne pas utiliser le dispositif de filtration pour le transfert de la solution filtrée de la seringue à la canule intraveineuse ou au set d'administration! |
Numéro d’autorisation
45217 (Swissmedic).
Titulaire de l’autorisation
Norgine AG, 6005 Lucerne
Mise à jour de l’information
Février 2021
Reviews (0)
Recently Viewed

Helixor A Injektionslösung 0.01mg Ampullen 8 Stück
Was ist Helixor und wann wird es angewendet?Gemäss der anthroposophischen Menschen- und Naturerkennt..
146.01 CHF

Free consultation with an experienced pharmacist
Describe the symptoms or the right drug - we will help you choose its dosage or analogue, place an order with home delivery or just consult.
We are 14 pharmacists and 0 bots. We will always be in touch with you and will be able to communicate at any time.
Bestsellers

Milupa Aptamil Pepti Syneo Can 400g
Product code: 7748334Aptamil Pepti Syneo is a special food for babies from birth who are allergic to cow's milk protein a..
55.08 CHF


Weleda Baby Calendula Wind- und Wetterbalsam 30ml
Product code: 5455461Der Weleda Baby Calendula Wind- und Wetterbalsam schützt die sensible Babyhaut intensiv bei Kälte un..
15.35 CHF


HerpoTherm herpes pen
Product code: 7798882Herpotherm® - the heating pen Cold sores are not only unattractive but can also be very painful. Th..
78.48 CHF



