




Ofev Kaps 100 mg of 60 pcs
Ofev Kaps 100 mg 60 Stk
-
3,505.65 CHF
- Price in reward points: 3131

- Availability: In stock
- Product Code: 6441917
- ATC-code L01EX09
- EAN 7680653300011
Ingredients:
Gelatine, Soja-Lecithin, Propylenglycol, Titandioxid (E171), Hartfett, Eisen(III)-oxid (E172), Triglyceride mittelkettige, Glycerol 85%, Schellack, Drucktinte schwarz, Nintedanib 100 mg , Nintedanib esilat 120.4 mg, Kapselhülle.

Description
 Deutsch
Deutsch French
French Italian
ItalianWas ist Ofev und wann wird es angewendet?
Ofev enthält den Wirkstoff Nintedanib. Es wird bei Erwachsenen zur Behandlung der idiopathischen Lungenfibrose (IPF) und anderer chronischer fibrosierender interstitieller Lungenerkrankungen (ILDs) mit progressivem Phänotyp sowie der mit systemischen Sklerose assoziierten interstitiellen Lungenerkrankung (SSc-ILD) angewendet und soll das Fortschreiten dieser Erkrankungen verlangsamen.
Idiopathische Lungenfibrose (IPF)
Bei der IPF kommt es mit der Zeit zu einer Verdickung, Versteifung und Vernarbung des Lungengewebes. Durch die Vernarbung kann nicht mehr genug Sauerstoff aus der Lunge in den Blutkreislauf transportiert werden und es kommt zu Atemproblemen. Ofev hilft, die Vernarbung und Versteifung der Lunge zu reduzieren.
Andere chronische fibrosierende interstitielle Lungenerkrankungen (ILDs) mit progressivem Phänotyp
Neben IPF gibt es weitere Erkrankungen, bei denen es zu einer Verdickung, Versteifung und mit der Zeit zu einer Vernarbung des Lungengewebes (Lungenfibrose) kommt und die sich weiter verschlimmern (progressiver Phänotyp). Dazu gehören Erkrankungen wie Hypersensitivitätspneumonitis, Autoimmun-ILD (z.B. mit rheumatoider Arthritis assoziierte ILD), idiopathische nicht-spezifische interstitielle Pneumonie, nicht klassifizierbare idiopathische interstitielle Pneumonie und andere ILD. Ofev hilft, die weitere Vernarbung und Versteifung der Lunge zu reduzieren.
Mit systemischer Sklerose assoziierte interstitielle Lungenerkrankung (SSc-ILD)
Die systemische Sklerose, auch bekannt als Sklerodermie (SSc), ist eine seltene chronische Autoimmunerkrankung des Bindegewebes in vielen Körperteilen. Sie kann zu einer Fibrosierung (Vernarbung und Versteifung) der Haut und der Lunge sowie weiterer innerer Organe führen. Ist das Lungengewebe von der Fibrosierung betroffen, spricht man von einer interstitiellen Lungenerkrankung (ILD). Daher wird das gesamte Krankheitsbild als SSc-ILD bezeichnet. Durch die Vernarbung kann nicht mehr genug Sauerstoff aus der Lunge in den Blutkreislauf transportiert werden und es kommt zu Atemproblemen. Ofev hilft, die weitere Vernarbung und Versteifung der Lunge zu reduzieren.
Wann darf Ofev nicht eingenommen werden?
Ofev darf nicht eingenommen werden,
- wenn Sie allergisch (überempfindlich) gegen Nintedanib, Erdnüsse oder Soja oder einen der sonstigen Bestandteile dieses Arzneimittels sind.
- wenn Sie schwanger sind.
Wann ist bei der Einnahme / Anwendung von Ofev Vorsicht geboten?
Informieren Sie bitte Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, bevor Sie Ofev einnehmen:
- Wenn Sie eine Lebererkrankung haben oder hatten. Bei Patienten mit geringem Körpergewicht (< 65kg), Asiaten und Frauen besteht ein höheres Risiko für Leberenzymerhöhungen. Bei Patienten mit diesen Risikofaktoren wird eine engmaschige Überwachung empfohlen.
- Wenn Sie Nierenprobleme haben oder jemals hatten.
- Wenn Sie Blutungsstörungen haben oder hatten.
- Wenn Sie blutverdünnende Arzneimittel anwenden um Blutgerinnsel zu verhindern.
- Wenn Sie eine Herzerkrankung haben oder hatten (z.B. Herzinfarkt)
- Wenn Sie kürzlich eine Operation hatten oder sich einer Operation unterziehen müssen. Nintedanib kann die Wundheilung beeinflussen. Unter Umständen wird die Behandlung mit Ofev unterbrochen. Ihr Arzt bzw. Ihre Ärztin wird entscheiden, wann die Behandlung mit Ofev wieder fortgesetzt werden kann.
- wenn Sie ein Aneurysma (Erweiterung und Schwächung einer Blutgefässwand) oder einen Einriss in einer Blutgefässwand haben oder hatten.
Bitte informieren Sie Ihren Arzt oder Ihre Ärztin sofort, wenn während der Einnahme dieses Arzneimittels folgendes auftritt:
- Wenn Sie Durchfall bekommen. Es ist sehr wichtig, den Durchfall bereits bei den ersten Anzeichen zu behandeln.
- Wenn Sie unter Erbrechen oder Übelkeit leiden.
- Wenn Sie starke Schmerzen in der Magengegend, Fieber oder Schüttelfrost haben, oder unter Übelkeit, Erbrechen, Blähungen oder Unterleibsschmerzen leiden. Dies könnten Anzeichen eines Darmdurchbruchs («gastrointestinale Perforation») sein.
- Wenn Sie Schmerzen, Schwellungen, eine Rötung oder ein Wärmegefühl in einem Arm oder Bein haben. Dies könnten Symptome eines Blutgerinnsels (Thrombose) in einer Ihrer Venen sein.
- Wenn ein Druckgefühl oder Schmerzen im Brustkorb (typischerweise auf der linken Körperseite), Schmerzen in Nacken, Kiefer, Schulter oder Arm, beschleunigter Herzschlag, Kurzatmigkeit, Übelkeit oder Erbrechen auftreten. Dies könnten Symptome eines Herzinfarkts sein.
- Wenn Sie eine starke Blutung haben.
Wechselwirkungen mit anderen Arzneimitteln
Ofev kann mit bestimmten anderen Arzneimitteln Wechselwirkungen haben.
Die folgenden Arzneimittel können die Wirkung von Ofev, verstärken und so das Risiko von Nebenwirkungen erhöhen:
- Arzneimittel gegen Pilzinfektionen.
- Arzneimittel gegen bakterielle Infektionen mit dem Wirkstoff Erythromycin.
Die folgenden Arzneimittel können die Wirksamkeit von Ofev vermindern:
- Arzneimittel zur Behandlung von Tuberkulose mit dem Wirkstoff Rifampicin,
- Arzneimittel zur Behandlung der Epilepsie,
- Arzneimittel mit Johanniskraut, pflanzliche Präparate gegen Depressionen.
Wenn Sie Raucher sind, sollten Sie das Rauchen während der Behandlung unterlassen, da dies die Wirksamkeit von Ofev verringern kann.
Aufgrund der Nebenwirkungen kann Ofev die Reaktionsfähigkeit, die Fahrtüchtigkeit und die Fähigkeit, Werkzeuge oder Maschinen zu bedienen, beeinträchtigen. Sie sollten keine Fahrzeuge lenken oder Maschinen bedienen, wenn Sie sich schlecht fühlen.
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wenn Sie
- an anderen Krankheiten leiden,
- Allergien haben oder
- andere Arzneimittel (auch selbst gekaufte!) einnehmen oder äusserlich anwenden!
Darf Ofev während einer Schwangerschaft oder in der Stillzeit eingenommen / angewendet werden?
Schwangerschaft
Während der Schwangerschaft dürfen Sie Ofev nicht einnehmen, da dieses Arzneimittel das ungeborene Kind schädigen kann. Vor Beginn der Behandlung müssen Sie einen Schwangerschaftstest machen, um sicher zu sein, dass Sie nicht schwanger sind.
Empfängnisverhütung
Frauen müssen während der Behandlung mit Ofev sowie mindestens drei Monaten nach Einnahme der letzten Kapsel eine hoch wirksame Verhütungsmethode anwenden. Sprechen Sie mit Ihrem Arzt resp. Ärztin, welche Verhütungsmethode für Sie geeignet ist.
Da derzeit nicht bekannt ist, ob Ofev die Wirksamkeit hormoneller Verhütungsmittel (wie Antibabypille, Hormonspirale, Depot-Spritze, Hormonimplantat, Verhütungspflaster) einschränkt, müssen Sie zusätzlich eine sogenannte Barrieremethode (z.B. Kondom) anwenden.
Falls Sie während der Behandlung mit Ofev schwanger werden oder eine Schwangerschaft vermuten, informieren Sie sofort Ihren Arzt bzw. Ärztin. Er/sie wird entscheiden, ob Sie die Behandlung fortführen sollen.
Stillzeit
Während der Behandlung mit Ofev dürfen Sie nicht stillen, da ein Risiko für das gestillte Kind nicht ausgeschlossen werden kann.
Wie verwenden Sie Ofev?
Nehmen Sie Ofev immer genau nach Absprache mit Ihrem Arzt oder Apotheker bzw. mit Ihrer Ärztin oder Apothekerin. Fragen Sie bei Ihrem Arzt oder Apotheker bzw. bei Ihrer Ärztin oder Apothekerin nach, wenn Sie sich nicht sicher sind.
Die empfohlene Dosis beträgt eine Kapsel Ofev à 150mg zweimal täglich mit dem Essen in einem Abstand von ungefähr 12 Stunden jeweils zur selben Tageszeit, beispielsweise eine Kapsel am Morgen und eine Kapsel am Abend.
Nehmen Sie die Kapseln mit einem Glas Wasser ein und schlucken Sie diese im Ganzen (sie dürfen nicht zerkaut oder zerdrückt werden).
Beenden Sie die Behandlung nicht ohne vorherige Rücksprache mit Ihrem Arzt bzw. mit Ihrer Ärztin. Es ist wichtig, dass Sie das Arzneimittel so lange einnehmen, wie es Ihr Arzt oder Ihre Ärztin verordnet hat.
Ändern Sie nicht von sich aus die verschriebene Dosierung. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. mit Ihrer Ärztin oder Apothekerin.
Wenn Sie eine grössere Menge von Ofev eingenommen haben, als Sie sollten
Benachrichtigen Sie sofort Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin.
Wenn Sie die Einnahme von Ofev vergessen haben
Nehmen Sie nicht die doppelte Dosis ein, wenn Sie die vorherige Einnahme vergessen haben. Nehmen Sie die nächste Dosis am nächsten geplanten Einnahmezeitpunkt in der Dosierung wie von Ihrem Arzt oder Apotheker bzw. Ihrer Ärztin oder Apothekerin empfohlen.
Anwendung bei Kindern und Jugendlichen
Die Anwendung und Sicherheit von Ofev bei Kindern und Jugendlichen unter 18 Jahren ist bisher nicht geprüft worden. Kinder und Jugendliche sollten Ofev daher nicht einnehmen.
Welche Nebenwirkungen kann Ofev haben?
Während der Behandlung mit Ofev müssen Sie insbesondere darauf achten, ob die nachfolgenden Nebenwirkungen bei Ihnen auftreten:
Durchfall
Der Durchfall kann zu einem Flüssigkeitsverlust und zu einem Verlust von für Ihren Körper wichtigen Salzen (Elektrolyten, wie Natrium oder Kalium) führen. Trinken Sie bei den ersten Anzeichen von Durchfall viel Flüssigkeit und benachrichtigen Sie umgehend Ihren Arzt bzw. Ihre Ärztin. Nehmen Sie baldmöglichst ein Arzneimittel gegen Durchfall, beispielsweise eines mit dem Wirkstoff Loperamid, ein.
Die folgenden weiteren Nebenwirkungen wurden während der Behandlung mit Ofev beabachtet:
Idiopathische Lungenfibrose (IPF)
Sehr häufig (betrifft mehr als einen von 10 Anwendern):
Durchfall, Übelkeit, Schmerzen im Unterleib, erhöhte Leberenzymwerte im Blut.
Häufig (betrifft 1 bis 10 von 100 Anwendern):
Erbrechen, Appetitverlust, Gewichtsverlust, Blutungen, Hautausschlag, Kopfschmerzen.
Gelegentlich (betrifft 1 bis 10 von 1000 Anwendern):
Bluthochdruck (Hypertonie), Gelbsucht, Entzündung der Bauchspeicheldrüse, schwerwiegende Leberschädigungen, niedrige Anzahl an Blutplättchen (Thrombozytopenie), Juckreiz (Pruritus), Darmdurchbruch («gastrointestinale Perforation»), Darmentzündung (Kolitis), Haarausfall (Alopezie).
Häufigkeit nicht bekannt
Erweiterung und Schwächung einer Blutgefässwand oder Einriss in einer Blutgefässwand (Aneurysmen und Arteriendissektionen), Nierenversagen.
Andere chronische fibrosierende interstitielle Lungenerkrankungen (ILD) mit progressivem Phänotyp
Sehr häufig (betrifft mehr als einen von 10 Anwendern):
Appetitverlust, Durchfall, Übelkeit, Erbrechen, Schmerzen im Unterleib, erhöhte Leberenzymwerte im Blut.
Häufig (betrifft 1 bis 10 von 100 Anwendern):
Blutungen, Bluthochdruck (Hypertonie), Gewichtsverlust, Kopfschmerzen, Hautausschlag, schwerwiegende Leberschädigungen.
Gelegentlich (betrifft 1 bis 10 von 1000 Anwendern):
Juckreiz (Pruritus), niedrige Anzahl an Blutplättchen (Thrombozytopenie), Entzündung der Bauchspeicheldrüse, Gelbsucht, Haarausfall (Alopezie), Darmentzündung (Kolitis), Nierenversagen.
Häufigkeit nicht bekannt:
Erweiterung und Schwächung einer Blutgefässwand oder Einriss in einer Blutgefässwand (Aneurysmen und Arteriendissektionen), Darmdurchbruch («gastrointestinale Perforation»).
Mit systemischer Sklerose assoziierte interstitielle Lungenerkrankung (SSc-ILD)
Sehr häufig (betrifft mehr als einen von 10 Anwendern):
Durchfall, Übelkeit, Erbrechen, Schmerzen im Unterleib, erhöhte Leberenzymwerte im Blut.
Häufig (betrifft 1 bis 10 von 100 Anwendern):
Blutungen, Bluthochdruck (Hypertonie), Appetitverlust, Gewichtsverlust, Kopfschmerzen.
Gelegentlich (betrifft 1 bis 10 von 1000 Anwendern):
Hautausschlag, Juckreiz (Pruritus), niedrige Anzahl an Blutplättchen (Thrombozytopenie), schwerwiegende Leberschädigungen, Darmentzündung (Kolitis), Nierenversagen.
Häufigkeit nicht bekannt:
Entzündung der Bauchspeicheldrüse, Gelbsucht, Erweiterung und Schwächung einer Blutgefässwand oder Einriss in einer Blutgefässwand (Aneurysmen und Arteriendissektionen), Darmdurchbruch («gastrointestinale Perforation»), Haarausfall (Alopezie).
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Dies gilt insbesondere auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Was ist ferner zu beachten?
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Lagerungshinweis
Ofev muss bei Raumtemperatur (15-25° C) gelagert werden und muss in der Originalverpackung um den Inhalt vor Feuchtigkeit zu schützen, und ausserhalb der Reichweite von Kindern aufbewahrt werden.
Weitere Hinweise
Weitere Auskünfte erteilt Ihnen Ihr Arzt oder Apotheker, bzw. Ihre Ärztin oder Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in Ofev enthalten?
Wirkstoffe
Der Wirkstoff ist Nintedanib.
1 Weichkapsel enthält 100 mg oder 150 mg Nintedanib als Nintedanib-Esilat.
Hilfsstoffe
- Kapselinhalt: mittelkettige Triglyceride, Hartfett, Lecithin (Soja) (E322)
- Kapselhülle: Gelatine, Glycerol (85 %), Titandioxid (E171), Eisenoxid gelb (E172), Eisenoxid rot (E172)
- Drucktinte: Schellack, Eisenoxid schwarz (E172), Propylenglycol (E1520)
Zulassungsnummer
65330 (Swissmedic)
Wo erhalten Sie Ofev? Welche Packungen sind erhältlich?
In Apotheken nur gegen ärztliche Verschreibung.
Ofev 100 mg: Packungen zu 60 Kapseln.
Ofev 150 mg: Packungen zu 60 Kapseln.
Zulassungsinhaberin
Boehringer Ingelheim (Schweiz) GmbH, Basel
Diese Packungsbeilage wurde im Oktober 2020 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
Qu’est-ce que Ofev et quand doit-il être utilisé?
Ofev contient le principe actif nintédanib. Il est utilisé chez les adultes pour le traitement de la fibrose pulmonaire idiopathique (FPI) et d'autres pneumopathies interstitielles (PI) fibrosantes chroniques avec un phénotype progressif, de même que la pneumopathie interstitielle liée à la sclérose systémique (PID-ScS) afin de ralentir la progression de ces maladies.
Fibrose pulmonaire idiopathique (FPI)
La FPI entraine un épaississement, une rigidification et la formation de cicatrices sur le tissu pulmonaire. A cause de ce tissu cicatriciel, l'oxygène des poumons n'est plus transporté en quantité suffisante dans la circulation sanguine, ce qui provoque des problèmes respiratoires. Ofev aide à réduire la formation de cicatrices et la rigidification des poumons.
Autres pneumopathies interstitielles (PI) fibrosantes chroniques avec un phénotype progressif
Outre la FPI, il existe d'autres affections qui entraînent un épaississement, une rigidification et, avec le temps, l'apparition de cicatrices au niveau du tissu pulmonaire (fibrose pulmonaire) qui se détériorent progressivement (phénotype progressif). Parmi celles-ci, on compte des affections telles que la pneumopathie d'hypersensibilité, la PI auto-immune (p.ex. PI liée à l'arthrite rhumatoïde), la pneumonie interstitielle non spécifique idiopathique, la pneumonie interstitielle idiopathique non classifiable et d'autres PI. Ofev aide à réduire la progression de l'apparition de cicatrices et de la rigidification des poumons.
Pneumopathie interstitielle liée à la sclérose systémique (PID-ScS)
La sclérose systémique, aussi appelée sclérodermie (ScS), est une maladie auto-immune chronique rare du tissu conjonctif dans de nombreuses parties du corps. Elle peut provoquer une fibrose (formation de cicatrices et rigidification) de la peau et des poumons, de même que d'autres organes internes. Si le tissu pulmonaire est touché par la fibrose, on parle de pneumopathie interstitielle (FPI). C'est pourquoi le tableau pathologique global est appelé PID-ScS. Le tissu cicatriciel ne permet plus le passage d'une quantité suffisante d'oxygène des poumons dans la circulation sanguine et entraîne des problèmes respiratoires. Ofev aide à réduire le tissu cicatriciel et la rigidité des poumons.
Quand Ofev ne doit-il pas être pris/utilisé?
Ofev ne doit pas être pris,
- si vous êtes allergique (hypersensible) au nintédanib, à l'arachide ou au soja, ou à l'un des autres composants de ce médicament.
- si vous êtes enceinte.
Quelles sont les précautions à observer lors de la prise/de l’utilisation de Ofev?
Avant de prendre Ofev, veuillez informer votre médecin ou votre pharmacien(ne) si
- si vous souffrez ou avez souffert d'une maladie du foie. Les patients à faible poids corporel (< 65 kg), les Asiatiques et les femmes présentent un risque plus élevé d'augmentations des taux d'enzymes hépatiques. Chez les patients présentant ces facteurs de risque, une surveillance étroite est recommandée.
- si vous avez ou avez déjà eu des problèmes rénaux.
- si vous souffrez ou avez souffert de troubles de la coagulation sanguine.
- si vous utilisez des anticoagulants pour éviter des caillots.
- si vous souffrez ou avez souffert d'une maladie du cœur (p.ex. crise cardiaque).
- si vous avez subi récemment une opération ou devez subir une opération d'ici peu. Le nintédanib peut influencer la cicatrisation. Le traitement par Ofev sera éventuellement arrêté. Votre médecin décidera quand reprendre le traitement par Ofev.
- si vous souffrez ou avez souffert d'un anévrisme (élargissement et affaiblissement de la paroi d'un vaisseau sanguin) ou d'une déchirure dans la paroi d'un vaisseau sanguin.
Veuillez informer directement votre médecin si les troubles suivants apparaissent au cours de la prise de ce médicament:
- si vous avez de la diarrhée. Il est très important de traiter la diarrhée de manière précoce.
- si vous souffrez de vomissements ou de nausées.
- si vous ressentez une douleur intense au niveau de l'estomac, si vous avez de la fièvre ou des frissons, des nausées, des vomissements, des flatulences ou des douleurs du bas ventre, car ces troubles pourraient indiquer une perforation gastro-intestinale.
- si vous ressentez une douleur, un gonflement, une rougeur ou de la chaleur dans un bras ou une jambe, car ces troubles pourraient être des symptômes d'un caillot sanguin (thrombose) dans une de vos veines.
- si vous ressentez une oppression ou une douleur au niveau de la poitrine (généralement du côté gauche du corps), une douleur dans le cou, la mâchoire, l'épaule ou le bras, si vous avez des palpitations cardiaques, si vous présentez un essoufflement, si vous avez des nausées ou des vomissements, car ces troubles pourraient être des symptômes d'une crise cardiaque.
- si vous présentez des saignements importants.
Interactions avec d'autres médicaments
Ofev peut interagir avec certains autres médicaments. Les médicaments suivants peuvent renforcer l'effet d'Ofev et donc augmenter le risque d'effets secondaires:
- médicaments contre les infections fongiques,
- médicament contre les infections bactériennes contenant le principe actif érythromycine.
Les médicaments suivants peuvent réduire l'effet d'Ofev:
- médicaments utilisés pour traiter la tuberculose contenant le principe actif rifampicine,
- médicaments utilisés pour traiter l'épilepsie,
- médicaments contenant du millepertuis, une plante médicinale utilisée pour traiter la dépression.
Si vous fumez, vous devriez arrêter de fumer pendant le traitement, car ceci peut réduire l'efficacité d'Ofev.
À cause des effets secondaires, Ofev peut affecter les réactions, l'aptitude à conduire et la capacité à utiliser des outils ou des machines. Vous ne devriez pas conduire ou utiliser des machines si vous vous sentez mal.
Veuillez informer votre médecin ou votre pharmacien si
- vous souffrez d'une autre maladie
- vous êtes allergique
- vous prenez déjà d'autres médicaments ou utilisez déjà d'autres médicaments en usage externe (même en automédication !).
Ofev peut-il être pris/utilisé pendant la grossesse ou l’allaitement?
Grossesse
Vous ne devez pas prendre Ofev pendant la grossesse, car ce médicament peut nuire à l'enfant à naitre. Avant de commencer le traitement, vous devez faire un test de grossesse afin de vous assurer que vous n'êtes pas enceinte.
Contraception
Les femmes doivent utiliser une contraception très efficace pendant le traitement par Ofev ainsi que trois mois au moins après la prise de la dernière capsule. Demandez conseil à votre médecin pour trouver une méthode de contraception qui vous convient.
Étant donné qu'on ignore encore si Ofev limite l'efficacité des méthodes de contraception hormonales (comme la pilule contraceptive, le stérilet hormonal, un contraceptif injectable, l'implant hormonal, le patch contraceptif), vous devez utiliser en plus une méthode dite de barrière (p.ex. préservatif). Informez immédiatement votre médecin si vous tombez enceinte ou vous soupçonnez une grossesse pendant le traitement par Ofev. Il décidera si vous pouvez continuer le traitement.
Allaitement
Vous ne devez pas allaiter pendant le traitement par Ofev, car un risque pour le nourrisson ne peut être exclu.
Comment utiliser Ofev?
Veillez à toujours prendre Ofev selon les indications de votre médecin ou pharmacien(ne). Vérifiez auprès de votre médecin ou pharmacien(ne) en cas de doute.
La dose recommandée est une capsule de 150 mg deux fois par jour avec les repas. Prenez les capsules à 12 heures d'intervalle, toujours approximativement à la même heure du jour (p.ex. une capsule le matin et une capsule le soir).
Prenez les capsules avec un verre d'eau et avalez-les entièrement (ne pas mâcher ou écraser).
N'arrêtez pas le traitement sans en avoir discuté avec votre médecin. Il est important que vous preniez le médicament aussi longtemps que le médecin vous l'a prescrit.
Ne changez pas de votre propre chef le dosage prescrit. Adressez-vous à votre médecin ou votre pharmacien(ne) si vous estimez que l'efficacité du médicament est trop faible ou au contraire trop forte.
Si vous avez pris plus d'Ofev que vous n'auriez dû
Contactez immédiatement votre médecin ou pharmacien(ne).
Si vous avez oublié de prendre Ofev
Ne prenez pas de double dose si vous avez oublié la prise précédente. Prenez la dose suivante au moment programmé suivant, avec le dosage tel que recommandé par votre médecin ou pharmacien(ne).
Utilisation chez les enfants et adolescents
L'utilisation et la sécurité d'Ofev n'ont pas été établies à ce jour pour les enfants et adolescents de moins de 18 ans. C'est pourquoi les enfants et adolescents ne doivent pas prendre Ofev.
Quels effets secondaires Ofev peut-il provoquer?
Pendant le traitement par Ofev, vous devez porter une attention particulière à la survenue des effets secondaires suivants:
Diarrhée
Une diarrhée peut entrainer une perte de liquide et de sels importants pour votre corps (électrolytes, tels le sodium ou le potassium) (déshydratation). Buvez beaucoup de liquides dès les premiers signes de diarrhée et contactez immédiatement votre médecin. Prenez dès que possible un médicament contre la diarrhée (antidiarrhéique), contenant par exemple du lopéramide.
Les effets indésirables suivants ont été observés pendant le traitement par Ofev:
Fibrose pulmonaire idiopathique (FPI)
Très fréquents (concerne plus de 10 utilisateurs)
Diarrhée, nausées, douleurs dans le bas ventre, valeurs d'enzymes hépatiques élevées dans le sang.
Fréquents (concerne de 1 à 10 utilisateurs sur 100)
Vomissements, perte d'appétit, perte de poids, hémorragies, éruptions cutanées, céphalées.
Occasionnels (concernent 1 à 10 utilisateurs sur 1000)
Hypertension, jaunisse, pancréatite, lésions hépatiques sévères, diminution du nombre de plaquettes (thrombocytopénie), démangeaisons (prurit), perforation gastro-intestinale, inflammation du côlon (colite), chute de cheveux (alopécie).
Fréquence inconnue
Élargissement et affaiblissement de la paroi d'un vaisseau sanguin ou déchirure dans la paroi d'un vaisseau sanguin (anévrismes et dissections artérielles), défaillance rénale.
Autres pneumopathies interstitielles (PI) fibrosantes chroniques avec un phénotype progressif
Très fréquents (concerne plus d'un utilisateur sur 10)
Perte d'appétit, diarrhée, nausées, vomissements, douleurs dans le bas-ventre, valeurs d'enzymes hépatiques élevées dans le sang.
Fréquents (concerne 1 à 10 utilisateurs sur 100)
Hémorragies, hypertension, perte de poids, céphalées, éruption cutanée, lésions hépatiques sévères.
Occasionnels (concerne 1 à 10 utilisateurs sur 1000)
Démangeaisons (prurit), diminution du nombre de plaquettes (thrombocytopénie), pancréatite, jaunisse, chute de cheveux (alopécie), inflammation intestinale (colite), défaillance rénale.
Fréquence inconnue
Elargissement et affaiblissement de la paroi d'un vaisseau sanguin ou déchirure dans la paroi d'un vaisseau sanguin (anévrismes et dissections artérielles), perforation gastro-intestinale.
Pneumopathie interstitielle liée à la sclérose systémique (PID-ScS)
Très fréquents (concerne plus d'un utilisateur sur 10)
Diarrhée, nausées, vomissements, douleurs dans le bas-ventre, valeurs d'enzymes hépatiques élevées dans le sang.
Fréquents (concerne de 1 à 10 utilisateurs sur 100)
Hémorragies, hypertension, perte d'appétit, perte de poids, céphalées.
Occasionnels (concerne 1 à 10 utilisateurs sur 1000)
Éruption cutanée, démangeaisons (prurit), dminution du nombre de plaquettes (thrombocytopénie), lésions hépatiques sévères, inflammation intestinale (colite), défaillance rénale.
Fréquence inconnue
Pancréatite, jaunisse, élargissement et affaiblissement de la paroi d'un vaisseau sanguin ou déchirure dans la paroi d'un vaisseau sanguin (anévrismes et dissections artérielles), perforation gastro-intestinale, chute de cheveux (alopécie).
Si vous remarquez des effets secondaires, veuillez en informer votre médecin ou votre pharmacien. Ceci vaut en particulier pour les effets secondaires non mentionnés dans cette notice d'emballage.
À quoi faut-il encore faire attention?
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques concernant le stockage
Ofev doit être conservé à température ambiante (15-25°C) et dans l'emballage d'origine afin de le protéger de l'humidité. Conserver hors de la portée des enfants.
Remarques complémentaires
Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien qui disposent d'une information détaillée destinée aux professionnels.
Que contient Ofev?
Principes actifs
Le principe actif est le nintédanib.
1 capsule molle contient 100 mg ou 150 mg de nintédanib sous forme d'ésilate de nintédanib.
Excipients
- Contenu des capsules: triglycérides à chaîne moyenne, graisse solide, lécithine (de soja) (E322)
- Enveloppe de la capsule: gélatine, glycérol (85 %), dioxyde de titane (E171), oxyde de fer jaune (E172), oxyde de fer rouge (E172)
- Encre d'impression: gomme laque, oxyde de fer noir (E172), propylèneglycol (E1520)
Numéro d’autorisation
65330 (Swissmedic)
Où obtenez-vous Ofev? Quels sont les emballages à disposition sur le marché?
En pharmacie, sur ordonnance médicale.
Ofev 100 mg: emballages de 60 capsules.
Ofev 150 mg: emballages de 60 capsules.
Titulaire de l’autorisation
Boehringer Ingelheim (Suisse) GmbH, Bâle
Cette notice d'emballage a été vérifiée pour la dernière fois en octobre 2020 par l'autorité de contrôle des médicaments (Swissmedic).
Che cos’è Ofev e quando si usa?
Ofev contiene il principio attivo nintedanib e viene utilizzato negli adulti per il trattamento della fibrosi polmonare idiopatica (IPF), di altre malattie interstiziali polmonari (ILD) fibrosanti croniche con fenotipo progressivo e della malattia interstiziale polmonare associata a sclerosi sistemica (SSc-ILD) allo scopo di rallentare la progressione delle malattie.
Fibrosi polmonare idiopatica (IPF)
L'IPF provoca con il tempo l'ispessimento, l'irrigidimento e la cicatrizzazione del tessuto polmonare. La cicatrizzazione impedisce all'ossigeno di essere trasportato in quantità adeguate dai polmoni nel circolo ematico, causando problemi di respirazione. Ofev aiuta a ridurre la cicatrizzazione e l'irrigidimento dei polmoni.
Altre malattie interstiziali polmonari (ILD) fibrosanti croniche con fenotipo progressivo
Oltre all'IPF esistono altre malattie che provocano un ispessimento, irrigidimento e, con il passare del tempo, una cicatrizzazione del tessuto polmonare (fibrosi polmonare) con graduale peggioramento (fenotipo progressivo). Ne fanno parte malattie come la polmone da ipersensibilità, l'ILD autoimmune (ad es. ILD associata ad artrite reumatoide), la polmonite interstiziale idiopatica non specifica, la polmonite interstiziale idiopatica non classificata e altre ILD. Ofev aiuta a ridurre la cicatrizzazione e l'irrigidimento dei polmoni.
Malattia interstiziale polmonare associata a sclerosi sistemica (SSc-ILD)
La sclerosi sistemica, nota anche come sclerodermia (SSc), è una rara malattia autoimmune cronica che colpisce il tessuto connettivo di diverse parti del corpo. La SSc causa fibrosi (cicatrizzazione e irrigidimento) della pelle, dei polmoni e di altri organi interni. Quando i polmoni sono colpiti da fibrosi, si parla di malattia interstiziale polmonare (ILD) e la patologia è detta SSc-ILD. La fibrosi riduce la capacità dei polmoni di trasportare ossigeno al sangue e la capacità respiratoria. Ofev contribuisce a ridurre l'ulteriore cicatrizzazione e irrigidimento dei polmoni.
Quando non si può assumere/usare Ofev?
Non deve assumere Ofev
- se è allergico (ipersensibile) al nintedanib, alle arachidi o alla soia o ad uno qualsiasi delle altre sostanze ausiliarie del medicamento.
- se è in stato di gravidanza.
Quando è richiesta prudenza nella somministrazione/nell’uso di Ofev?
Informi il suo medico o il suo farmacista prima di assumere Ofev:
- se soffre o ha sofferto di una malattia epatica. Nei pazienti con peso corporeo ridotto (< 65 kg), negli asiatici e nelle donne sussiste un maggior rischio di aumento dei livelli degli enzimi epatici. Si raccomanda uno stretto monitoraggio dei pazienti con questi fattori di rischio.
- se soffre o ha mai avuto in passato, problemi renali.
- se soffre o ha sofferto di disturbi emorragici.
- se fa uso di anticoagulanti per evitare la formazione di coaguli di sangue.
- se soffre o ha sofferto di una malattia cardiaca (p. es. infarto del miocardio).
- se si è sottoposto di recente o sta per sopporsi ad un intervento chirurgico. Nintedanib può influenzare il processo di guarigione delle ferite. Il trattamento con Ofev potrebbe essere interrotto. Spetterà al suo medico decidere quando sarà opportuno riprendere il trattamento con Ofev.
- se ha o ha avuto un aneurisma (dilatazione e indebolimento della parete di un vaso sanguigno) o una lacerazione della parete di un vaso sanguigno.
Mentre prende questo medicamento informi subito il suo medico:
- se ha la diarrea. È molto importante trattare la diarrea fin dalla comparsa dei primi segni.
- se soffre di vomito o nausea.
- se accusa forti dolori nella regione addominale, ha febbre o brividi o soffre di nausea, vomito, flatulenza, o dolori al basso ventre. Potrebbero essere segni di perforazione gastrointestinale (un'apertura nella parete dello stomaco/intestino).
- se ha dolore, gonfiore, arrossamento o prova una sensazione di calore a un braccio o a una gamba. Potrebbero essere sintomi della formazione di un coagulo di sangue (trombosi) in una vena.
- se sente pressione o dolore al petto (particolarmente sul lato sinistro del corpo), dolore al collo, alla mandibola, alla spalla o al braccio, se presenta battito cardiaco accelerato, dispnea, nausea o vomito. Potrebbero essere i sintomi di un infarto del miocardio.
- se ha una forte emorragia.
Interazioni con altri medicamenti
Ofev può interagire con determinati medicamenti. I seguenti medicamenti possono rinforzare l'azione di Ofev e quindi aumentare il rischio di effetti collaterali:
- medicamenti utilizzati per le infezioni fungine.
- medicamenti utilizzati per le infezioni batteriche contenenti il principio attivo eritromicina.
I seguenti medicamenti possono ridurre l'efficacia di Ofev:
- antitubercolotici contenenti il principio attivo rifampicina,
- antiepilettici,
- medicamenti contenenti iperico (erba di San Giovanni), rimedi fitoterapici per la depressione.
Se fuma, durante il trattamento dovrebbe smettere di fumare per non compromettere l'efficacia di Ofev.
A causa degli effetti collaterali, Ofev può ridurre la capacità di reazione, la capacità di condurre un veicolo e la capacità di utilizzare attrezzi e macchine. Non guidi né azioni macchinari se non si sente bene.
Informi il suo medico o il suo farmacista, nel caso in cui
- soffre di altre malattie
- soffre di allergie o
- assume altri medicamenti (anche se acquistati di sua iniziativa) o li applica esternamente
Si può assumere/usare Ofev durante la gravidanza o l’allattamento?
Gravidanza
Non assuma Ofev durante la gravidanza poiché questo medicamento può danneggiare il feto. È necessario che si sottoponga a un test di gravidanza prima dell'inizio del trattamento per escludere un'eventuale gravidanza.
Contraccezione
Le donne devono adottare un metodo contraccettivo altamente efficace durante il trattamento con Ofev e almeno per i tre mesi successivi all'assunzione dell'ultima capsula del medicamento. Discuta con il suo medico il metodo anticoncezionale più indicato nel suo caso.
Attualmente non è noto se Ofev riduca o meno l'efficacia dei contraccettivi ormonali (come pillola anticoncezionale, spirale ormonale, iniezione a deposito, impianto ormonale, cerotto anticoncezionale), pertanto deve usare anche un metodo contraccettivo barriera (ad es. profilattico).
Se dovesse rimanere incinta o sospettasse di essere rimasta incinta durante il trattamento con Ofev, informi immediatamente il suo medico o il suo farmacista, cui spetterà decidere se farla continuare o meno il trattamento.
Allattamento
Non allatti durante il trattamento con Ofev poiché non è possibile escludere un rischio per il bambino allattato al seno.
Come usare Ofev?
Assuma Ofev seguendo sempre attentamente le indicazioni del suo medico o del suo farmacista. In caso di dubbi, si rivolga al suo medico o al suo farmacista.
La dose raccomandata di Ofev è di una capsula da 150 mg due volte al giorno da assumere con il cibo ad intervalli di circa 12 ore, sempre alla stessa ora, ad esempio una capsula alla mattina e una alla sera.
Prenda le capsule con un bicchiere d'acqua e le ingoi intere (non masticare o frantumare le capsule).
Non interrompa il trattamento senza aver prima consultato il suo medico. È importante che continui ad assumere il medicamento per tutta la durata indicatale dal suo medico.
Non modifichi di propria iniziativa la posologia prescritta. Se ritiene che l'azione del medicamento sia troppo debole o troppo forte, ne parli al suo medico o al suo farmacista.
Se prende più Ofev di quanto deve
Si rivolga immediatamente al suo medico o al suo farmacista.
Se dimentica di prendere Ofev
Non assuma una dose doppia se ha dimenticato di prendere la dose precedente. Prenda la dose successiva all'orario successivo previsto come programmato, e alla dose raccomandata dal suo medico o dal suo farmacista.
Uso nei bambini e negli adolescenti
L'uso e la sicurezza di Ofev nei bambini e negli adolescenti sotto i 18 anni finora non sono stati esaminati. I bambini e gli adolescenti non devono quindi assumere Ofev.
Quali effetti collaterali può avere Ofev?
Durante il trattamento con Ofev deve prestare particolare attenzione se manifesta i seguenti effetti collaterali:
Diarrea
La diarrea può causare disidratazione e perdita di sali importanti per l'organismo (elettroliti quali sodio o potassio). Beva molti liquidi ai primi segni di diarrea e informi immediatamente il suo medico o il suo farmacista. Prenda quanto prima un antidiarroico, ad esempio un medicamento contenente il principio attivo loperamide.
Durante il trattamento con Ofev sono stati osservati anche i seguenti effetti collaterali:
Fibrosi polmonare idiopatica (IPF)
Molto comune (riguarda più di 1 utilizzatore su 10):
Diarrea, nausea, dolori al basso ventre, aumento degli enzimi epatici nel sangue.
Comune (riguarda da 1 a 10 utilizzatori su 100):
Vomito, inappetenza, diminuzione del peso, sanguinamenti, eruzione cutanea, mal di testa.
Non comune (riguarda da 1 a 10 utilizzatori su 1000):
Pressione arteriosa elevata (ipertensione), ittero, infiammazione del pancreas, gravi lesioni epatiche, ridotta quantità di piastrine nel sangue (trombocitopenia), prurito, perforazione gastrointestinale, infiammazione dell'intestino (colite), perdita dei capelli (alopecia).
Frequenza non nota:
Dilatazione e indebolimento della parete di un vaso sanguigno o una lacerazione della parete di un
vaso sanguigno (aneurismi e dissezioni arteriose), insufficienza renale.
Altre malattie interstiziali polmonari (ILD) fibrosanti croniche con fenotipo progressivo
Molto comune (riguarda più di 1 utilizzatore su 10):
Inappetenza, diarrea, nausea, vomito, dolori al basso ventre, aumento degli enzimi epatici nel sangue.
Comune (riguarda da 1 a 10 utilizzatori su 100):
Sanguinamenti, pressione arteriosa elevata (ipertensione), diminuzione del peso, mal di testa, eruzione cutanea, gravi lesioni epatiche.
Non comune (riguarda da 1 a 10 utilizzatori su 1000):
Prurito, ridotta quantità di piastrine nel sangue (trombocitopenia), infiammazione del pancreas, ittero, perdita dei capelli (alopecia), infiammazione dell'intestino (colite), insufficienza renale.
Frequenza non nota:
Dilatazione e indebolimento della parete di un vaso sanguigno o una lacerazione della parete di un vaso sanguigno (aneurismi e dissezioni arteriose), perforazione gastrointestinale.
Malattia inerstiziale polmonare associata a sclerosi sistemica (SSc-ILD)
Molto comune (riguarda più di 1 utilizzatore su 10):
Diarrea, nausea, vomito, dolori al basso ventre, aumento degli enzimi epatici nel sangue.
Comune (riguarda da 1 a 10 utilizzatori su 100):
Sanguinamenti, pressione arteriosa elevata (ipertensione), inappetenza, diminuzione del peso, mal di testa.
Non comune (riguarda da 1 a 10 utilizzatori su 1000):
Eruzione cutanea, prurito, ridotta quantità di piastrine nel sangue (trombocitopenia), gravi lesioni epatiche, infiammazione dell'intestino (colite), insufficienza renale.
Frequenza non nota:
Infiammazione del pancreas, ittero, dilatazione e indebolimento della parete di un vaso sanguigno o lacerazione della parete di un vaso sanguigno (aneurismi e dissezioni arteriose), perforazione gastrointestinale, perdita dei capelli (alopecia).
Se osserva effetti collaterali, si rivolga al suo medico o farmacista, soprattutto se si tratta di effetti collaterali non descritti in questo foglietto illustrativo.
Di che altro occorre tener conto?
Il medicamento non dev'essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Indicazione di stoccaggio
Ofev deve essere conservato a temperatura ambiente (15-25° C) e va tenuto nella confezione originale per proteggere il contenuto dall'umidità e fuori dalla portata dei bambini.
Ulteriori indicazioni
Il medico o il farmacista, che sono in possesso di un'informazione professionale dettagliata, possono darle ulteriori informazioni.
Cosa contiene Ofev?
Principi attivi
Il principio attivo è nintedanib.
1 capsula molle contiene 100 mg o 150 mg di nintedanib sotto forma di nintedanib esilato.
Sostanze ausiliarie
- Contenuto della capsula: trigliceridi a catena media, grasso solido, lecitina (soia) (E322)
- Capsula: gelatina, glicerolo (85%), titanio diossido (E171), ferro ossido giallo (E172), ferro ossido rosso (E172)
- Inchiostro: gomma lacca, ferro ossido nero (E172), glicole propilenico (E1520)
Numero dell’omologazione
65330 (Swissmedic)
Dove è ottenibile Ofev? Quali confezioni sono disponibili?
In farmacia, dietro presentazione della prescrizione medica.
Ofev 100 mg: confezioni da 60 capsule.
Ofev 150 mg: confezioni da 60 capsule.
Titolare dell’omologazione
Boehringer Ingelheim (Schweiz) GmbH, Basel
Questo foglietto illustrativo è stato controllato l'ultima volta nell'ottobre 2020 dall'autorità competente in materia di medicamenti (Swissmedic).
Zusammensetzung
Wirkstoffe
Nintedanib (als Nintedanib-Esilat).
Hilfsstoffe
- Kapselinhalt: mittelkettige Triglyceride, Hartfett, Lecithin (Soja)(E322)
- Kapselhülle: Gelatine, Glycerol (85 %), Titandioxid (E171), Eisenoxid gelb (E172), Eisenoxid rot (E172)
- Drucktinte: Schellack, Eisenoxid schwarz (E172), Propylenglycol (E1520)
Darreichungsform und Wirkstoffmenge pro Einheit
Weichkapseln zu 100 mg und 150 mg Nintedanib (entsprechend 120,40 mg und 180,60 mg Nintedanib-Esilat)
Indikationen/Anwendungsmöglichkeiten
Ofev ist indiziert für die Behandlung
- der idiopathischen Lungenfibrose (IPF)
- von chronischen fibrosierenden interstitiellen Lungenerkrankungen (ILDs) mit einem progressiven Phänotyp (siehe Rubrik «Klinische Wirksamkeit»)
- der mit systemischer Sklerose assoziierten interstitiellen Lungenerkrankung (SSc-ILD)
Dosierung/Anwendung
Die Behandlung sollte von einem Arzt eingeleitet werden, der Erfahrung mit der Diagnostik und Therapie der Erkrankungen, für die Ofev indiziert ist, hat.
Die empfohlene Dosis von Ofev beträgt 150 mg zweimal täglich im Abstand von etwa 12 Stunden.
Ofev Kapseln sollen im Ganzen mit Wasser und zu Nahrung eingenommen werden. Die Kapseln dürfen nicht zerkaut oder anderweitig zerkleinert werden.
Wenn eine Dosis Ofev ausgelassen wurde, die Behandlung am nächsten geplanten Einnahmezeitpunkt in empfohlener Dosierung fortsetzen. Wenn eine Dosis ausgelassen wurde, sollte der Patient keine zusätzliche Dosis erhalten. Die empfohlene maximale Tagesdosis von 300 mg sollte nicht überschritten werden.
Dosierungsanpassungen
Massnahmen bei Nebenwirkungen (siehe Abschnitte «Warnhinweise und Vorsichtsmassnahmen», «Unerwünschte Wirkungen») von Ofev können zusätzlich zu einer symptomatischen Therapie (sofern angezeigt), eine Dosisreduktion oder eine vorübergehende Unterbrechung der Behandlung umfassen, bis die jeweilige Nebenwirkung abgeklungen ist oder auf einen Grad abgenommen hat, der eine Fortsetzung der Therapie erlaubt. Die Behandlung mit Ofev kann in voller Dosis (150 mg zweimal täglich) oder in verringerter Dosis (100 mg zweimal täglich) wieder aufgenommen werden. Verträgt der Patient eine Dosis von 100 mg zweimal täglich nicht, so sollte die Behandlung mit Ofev abgesetzt werden.
Bei Unterbrechung der Behandlung aufgrund eines Transaminasenanstiegs (AST oder ALT) auf mehr als das 3-fache der Obergrenze des Normbereichs (OGN) kann die Behandlung mit Ofev nach Normalisierung der Transaminasenwerte in verringerter Dosis (100 mg zweimal täglich) wieder aufgenommen und anschliessend wieder auf die volle Dosis (150 mg zweimal täglich) erhöht werden. Bei einem Anstieg der AST oder ALT auf mehr als das 5-fache der OGN oder bei einem Anstieg auf mehr als das 3-fache der OGN mit gleichzeitig bestehenden Befunden oder Symptomen einer schweren Leberschädigung ist Ofev abzusetzen (siehe «Warnhinweise und Vorsichtsmassnahmen», «Unerwünschte Wirkungen»).
Spezielle Dosierungsanweisungen
Patienten mit Nierenfunktionsstörungen
Weniger als 1 % einer Einzeldosis Nintedanib wird über die Nieren ausgeschieden (siehe «Pharmakokinetik»). Bei Patienten mit leichter bis mittelschwerer Einschränkung der Nierenfunktion ist keine Anpassung der Anfangsdosis erforderlich. Die Sicherheit, Wirksamkeit und Pharmakokinetik von Nintedanib bei Patienten mit schwerer Einschränkung der Nierenfunktion wurden nicht untersucht (CrCL <30 ml/min).
Patienten mit Leberfunktionsstörungen
Nintedanib wird vorwiegend über die Galle/die Fäzes ausgeschieden (> 90 %; siehe «Pharmakokinetik»). Die Exposition stieg bei Patienten mit Einschränkung der Leberfunktion (Child-Pugh-Klasse A, Child-Pugh-Klasse B) an. Bei Patienten mit leichter Beeinträchtigung der Leberfunktion (Child-Pugh-Klasse A) beträgt die empfohlene Ofev-Dosis 100 mg zweimal täglich im Abstand von jeweils etwa 12 Stunden. Bei Patienten mit leichter Beeinträchtigung der Leberfunktion (Child-Pugh-Klasse A) sollte eine Unterbrechung oder ein Absetzen der Behandlung, um unerwünschte Reaktionen behandeln zu können, in Erwägung gezogen werden.
Die Sicherheit und Wirksamkeit von Nintedanib bei Patienten mit Einschränkung der Leberfunktion vom Grad Child Pugh B oder C wurde nicht untersucht. Die Anwendung von Ofev bei Patienten mit mittelschwerer (Child-Pugh-Klasse B) oder schwerer (Child-Pugh-Klasse C) Einschränkung der Leberfunktion wird nicht empfohlen (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakokinetik»).
Kinder und Jugendliche
Die Sicherheit und Wirksamkeit von Ofev bei Kindern und Jugendlichen wurde nicht in klinischen Studien untersucht.
Ältere Patienten (≥65 Jahre)
Bei älteren Patienten wurden hinsichtlich der allgemeinen Sicherheit und Wirksamkeit keine Unterschiede zu Patienten unter 65 Jahren beobachtet. Es ist keine Dosisanpassung auf Grundlage des Lebensalters des Patienten erforderlich (siehe «Pharmakokinetik»).
Kontraindikationen
Ofev ist bei Patienten mit bekannter Überempfindlichkeit gegen Nintedanib, Erdnüsse, Soja oder einen der sonstigen Bestandteile kontraindiziert.
Ofev ist in der Schwangerschaft kontraindiziert (siehe «Schwangerschaft /Stillzeit» und «Präklinische Daten»).
Warnhinweise und Vorsichtsmassnahmen
Erkrankungen des Gastrointestinaltrakts
Diarrhö
In den klinischen Studien (siehe «Klinische Wirksamkeit») war Diarrhö das am häufigsten genannte gastrointestinale Ereignis.
Bei den meisten Patienten war Diarrhö leicht bis mittelschwer ausgeprägt und trat in den ersten 3 Behandlungsmonaten auf.
In den INPULSIS-Studien (IPF-Patienten) wurde eine Diarrhö bei 62,4% der mit Ofev versus 18,4% der mit Placebo behandelten Patienten berichtet. Durchfall führte bei 10,7% der Patienten zu einer Dosisreduktion von Ofev versus bei keinem mit Placebo behandelten Patienten und bei 4,4% der Patienten zum Abbruch der Behandlung mit Ofev versus 0,2% der mit Placebo behandelten Patienten.
In der INBUILD-Studie (Patienten mit progressiven fibrosierenden ILDs) wurde Diarrhö bei 66,9 % der mit Ofev versus 23,9 % der mit Placebo behandelten Patienten berichtet. Diarrhö führte bei 16,0 % der Patienten zu einer Dosisreduktion von Ofev versus 0,9% bei mit Placebo behandelten Patienten und bei 5,7 % der Patienten zum Abbruch der Behandlung mit Ofev versus 0,3% bei mit Placebo behandelten Patienten.
In der SENSCIS-Studie (SSc-ILD-Patienten) wurde Diarrhö bei 75,7 % der mit Ofev versus 31,6% der mit Placebo behandelten Patienten berichtet.
Durchfall führte bei 22,2% der Patienten zu einer Dosisreduktion von Ofev versus 1,0% bei mit Placebo behandelten Patienten und bei 6,9% der Patienten zum Abbruch der Behandlung versus 0,3% bei mit Placebo-behandelten Patienten (siehe Abschnitt «Unerwünschte Wirkungen»).
Diarrhö kann zu Dehydrierung mit oder ohne Elektrolytstörungen führen, was eine Nierenfunktionsstörung zur Folge haben kann.
Eine Diarrhö sollte bei den ersten Anzeichen mit angemessener Hydrierung und Antidiarrhoika wie beispielsweise Loperamid behandelt werden. Gegebenenfalls ist eine Dosisreduktion oder eine Unterbrechung der Behandlung erforderlich. Die Behandlung mit Ofev kann in verringerter Dosis (100 mg zweimal täglich) oder in voller Dosis (150 mg zweimal täglich) wieder aufgenommen werden. Wenn eine schwere Diarrhö trotz symptomatischer Therapie persistiert, sollte die Behandlung mit Ofev abgebrochen werden.
Übelkeit und Erbrechen
Übelkeit und Erbrechen sind häufig genannte unerwünschte Ereignisse (siehe «Unerwünschte Wirkungen») und meist leicht bis mittelschwer ausgeprägt.
In den INPULSIS-Studien (IPF-Patienten) wurde Übelkeit bei 24,5 % der mit Ofev versus 6,6 % der mit Placebo behandelten Patienten berichtet. Übelkeit führte bei 1,7 % der Patienten zu einer Dosisreduktion von Ofev versus bei keinem der mit Placebo behandelten Patienten und bei 2,0 % der Patienten zum Abbruch der Behandlung mit Ofev versus bei keinem der mit Placebo behandelten Patienten.
In der INBUILD-Studie (Patienten mit progressiven fibrosierenden ILDs) wurde Übelkeit bei 28,9 % der mit Ofev versus 9,4 % der mit Placebo behandelten Patienten berichtet. Übelkeit führte bei 3,3 % der Patienten zu einer Dosisreduktion von Ofev versus 0,6 % bei mit Placebo behandelten Patienten und bei 0,3 % der Patienten zum Abbruch der Behandlung mit Ofev versus 0,3 % bei mit Placebo behandelten Patienten.
In der SENSCIS-Studie (SSc-ILD-Patienten) wurde Übelkeit bei 31,6 % der mit Ofev versus 13,5 % der mit Placebo behandelten Patienten berichtet. Übelkeit führte bei 2,1 % der Patienten zu einer Dosisreduktion von Ofev versus bei keinem der mit Placebo behandelten Patienten und bei 2,1 % der Patienten zum Abbruch der Behandlung mit Ofev versus bei keinem der mit Placebo behandelten Patienten.
In den INPULSIS-Studien (IPF-Patienten) wurde Erbrechen bei 11,6 % der mit Ofev versus 2,6 % der mit Placebo behandelten Patienten berichtet. Erbrechen führte bei 1,1 % der Patienten zu einer Dosisreduktion von Ofev versus bei keinem der mit Placebo behandelten Patienten und bei 0,8 % der Patienten zum Abbruch der Behandlung mit Ofev versus bei keinem der mit Placebo behandelten Patienten.
In der INBUILD-Studie (Patienten mit progressiven fibrosierenden ILDs) wurde Erbrechen bei 18,4 % der mit Ofev versus 5,1 % der mit Placebo behandelten Patienten berichtet. Erbrechen führte bei 2,4 % der Patienten zu einer Dosisreduktion von Ofev versus 0,9 % bei mit Placebo behandelten Patienten und bei 0,9 % der Patienten zum Abbruch der Behandlung mit Ofev versus bei keinem der mit Placebo behandelten Patienten.
In der SENSCIS-Studie (SSc-ILD-Patienten) wurde Erbrechen bei 24,7 % der mit Ofev versus 10,4 % der mit Placebo behandelten Patienten berichtet. Erbrechen führte bei 2,1 % der Patienten zu einer Dosisreduktion von Ofev versus bei keinem der mit Placebo behandelten Patienten und bei 1,4 % der Patienten zum Abbruch der Behandlung mit Ofev versus 0,3 % bei mit Placebo behandelten Patienten.
Erbrechen kann zu Dehydrierung mit oder ohne Elektrolytstörungen führen, was eine Nierenfunktionsstörung zur Folge haben kann.
Wenn die Symptome trotz geeigneter supportiver Massnahmen (einschliesslich einer antiemetischen Therapie) persistieren, kann eine Dosisreduktion oder Unterbrechung der Behandlung erforderlich sein. Die Behandlung kann in verringerter Dosis (100 mg zweimal täglich) oder in voller Dosis (150 mg zweimal täglich) wieder aufgenommen werden. Bei persistierenden schweren Symptomen sollte die Behandlung mit Ofev abgesetzt werden.
Leberfunktion
Die Sicherheit und Wirksamkeit von Ofev bei Patienten mit mittelschwerer (Child-Pugh-Klasse B) oder schwerer (Child-Pugh-Klasse C) Einschränkung der Leberfunktion wurde nicht untersucht. Daher wird die Anwendung von Ofev bei diesen Patienten nicht empfohlen.
Aufgrund der erhöhten Exposition kann das Risiko für unerwünschte Ereignisse bei Patienten mit leichter Einschränkung der Leberfunktion (Child-Pugh-Klasse A) erhöht sein. Patienten mit leichter Beeinträchtigung der Leberfunktion (Child-Pugh-Klasse A) sollten mit einer reduzierten Ofev-Dosis behandelt werden (siehe «Dosierung / Anwendung» und «Pharmakokinetik»).
Fälle von arzneimittelinduzierten Leberschäden wurden unter der Behandlung mit Nintedanib beobachtet. In der Marktbeobachtung wurden nicht-schwerwiegende und schwerwiegende Fälle von arzneimittelinduzierten Leberschäden gemeldet, darunter auch schwere Leberschäden mit tödlichem Ausgang.
Die meisten unerwünschten hepatischen Ereignisse traten innerhalb der ersten drei Behandlungsmonate auf. Daher sollten die hepatischen Transaminasen und Bilirubin-Konzentrationen zu Beginn der Behandlung mit Ofev, in regelmässigen Abständen während der ersten drei Monate der Behandlung und anschliessend periodisch (z.B. bei jedem Termin des Patienten) oder wenn klinisch indiziert kontrolliert werden.
Erhöhungen der Leberenzymwerte (ALT, AST, ALKP, Gammaglutamyltransferase (GGT)) und des Bilirubinwerts waren nach Dosisreduktion oder Behandlungsunterbrechung in den meisten Fällen reversibel. Erhöhte ALT- und/oder AST-Werte, die auf mindestens das 3-Fache der OGN anstiegen, wurden bei bis zu 5,0%, 7,8% bzw. 4,9 % der mit Ofev behandelten Patienten in den INPULSIS (IPF-Patienten) -, INBUILD (Patienten mit progressiven fibrosierenden ILDs) - bzw. SENSCIS (SSc-ILD-Patienten) -Studien beobachtet, während es bei Patienten, die Placebo erhielten, insgesamt weniger als 2% waren.
Bei einem Anstieg der Transaminasen (AST oder ALT) auf das mehr als 3-fache der Obergrenze des Normbereichs (OGN) sollte die Behandlung mit Ofev unterbrochen oder die Dosis auf 100 mg zweimal täglich verringert werden. Ausserdem sollte der Patient engmaschig überwacht werden und alternative Ursachen für den Anstieg der Leberenzyme sind zu untersuchen. Nach Normalisierung der Transaminasen kann die Behandlung mit Ofev in verringerter Dosis (100 mg zweimal täglich) wieder aufgenommen und anschliessend auf die volle Dosis (150 mg zweimal täglich) erhöht werden. Bestehen parallel zu einem Anstieg von Leberwerten klinische Zeichen einer Leberschädigung wie beispielsweise eine Gelbsucht, oder steigen die Transaminasen (AST oder ALT) auf das mehr als 5-fache der OGN an, so ist die Behandlung mit Ofev endgültig abzusetzen (siehe «Dosierung/Anwendung»).
Bei Patienten mit geringem Körpergewicht (< 65 kg), Asiaten und Frauen besteht ein höheres Risiko für Leberenzymerhöhungen.
Die Nintedanib-Exposition erhöhte sich linear mit dem Alter der Patienten. Dies könnte auch zu einem Anstieg des Risikos für Leberenzymerhöhungen führen (siehe Abschnitt «Pharmakokinetik»).
Bei Patienten mit diesen Risikofaktoren wird eine engmaschige Überwachung empfohlen.
Nierenfunktion
Bei der Anwendung von Nintedanib wurde über Fälle von Nierenfunktionsstörungen bzw. Nierenversagen berichtet, von denen einige tödlich verliefen (siehe «Unerwünschte Wirkungen»).
Während der Therapie mit Nintedanib sollten die Patienten überwacht werden, insbesondere solche Patienten, die Risikofaktoren für eine Nierenfunktionsstörung bzw. ein Nierenversagen aufweisen. Bei einer Nierenfunktionsstörung bzw. einem Nierenversagen ist eine Anpassung der Therapie in Erwägung zu ziehen (siehe «Dosierungsanpassungen»).
Blutungen
Angesichts seines Wirkmechanismus (Hemmung des vaskulären endothelialen Wachstumsfaktorrezeptors (vascular endothelial growth factor receptor, VEGFR)) kann Ofev mit einem erhöhten Blutungsrisiko verbunden sein.
In klinischen Studien mit Ofev war die Häufigkeit von Blutungsereignissen bei Patienten unter Ofev etwas höher bzw. zwischen den Behandlungsgruppen vergleichbar (10,3% versus Placebo 7,8% in den INPULSIS-Studien (IPF-Patienten); 11,1 % versus Placebo 12,7% in der INBUILD-Studie (Patienten mit progressiven fibrosierenden ILDs),11,1 % versus Placebo 8,3% in der SENSCIS-Studie (SSc-ILD-Patienten)).
Nicht schwerwiegende Epistaxis stellte das häufigste Blutungsereignis dar.
Die Häufigkeit von schwerwiegenden Blutungsereignissen war in den beiden Behandlungsgruppen gering (Ofev 1,3 % versus Placebo 1,4 % in INPULSIS (IPF-Patienten); Ofev 0,9 % versus Placebo 1,5 % in INBUILD (Patienten mit progressiven fibrosierenden ILDs); Ofev 1,4 % versus Placebo 0,7 % in SENSCIS (SSc-ILD-Patienten))
An den klinischen-Studien nahmen keine Patienten mit bekanntem Blutungsrisiko wie solche mit angeborener Blutungsneigung oder Patienten, die eine Antikoagulation in voller Dosis erhalten, teil. Nach Markteinführung wurden Fälle von Blutungen berichtet, darunter schwerwiegende und tödliche (einschliesslich Patienten mit oder ohne Behandlung mit Antikoagulanzien oder anderen Arzneimitteln, die Blutungen hervorrufen könnten). Daher sollten Patienten mit zusätzlichen Risikofaktoren nur dann mit Ofev behandelt werden, wenn der erwartete Nutzen das potenzielle Risiko überwiegt.
Arterielle thromboembolische Ereignisse
Patienten mit kürzlichem Myokardinfarkt oder Schlaganfall waren von den klinischen Studien ausgeschlossen.
In den klinischen Studien wurden arterielle thromboembolische Ereignisse mit geringer Häufigkeit beschrieben (Ofev 2,5 % versus Placebo 0,7 % in INPULSIS (IPF-Patienten); Ofev 0,9 % versus Placebo 0,9 % in INBUILD (Patienten mit progressiven fibrosierenden ILDs); Ofev 0,7 % versus Placebo 0,7 % in SENSCIS (SSc-ILD-Patienten)). In den INPULSIS-Studien wurden bei einem höheren Prozentsatz der Patienten in der Ofev-Gruppe (1,6%) als der Patienten in der Placebogruppe (0,5 %) Myokardinfarkte beschrieben, während unerwünschte Ereignisse, die eine ischämische Herzkrankheit widerspiegeln, in der Ofev- und Placebogruppe ausgeglichen waren. In der INBUILD- und der SENSCIS-Studie wurde eine geringe Häufigkeit von Myokardinfarkten beobachtet: Ofev 0,9 % versus Placebo 0,9 % in INBUILD; Ofev 0 % versus Placebo 0,7 % in SENSCIS.
Bei Patienten mit erhöhtem kardiovaskulärem Risiko, wie beispielsweise solchen mit bekannter koronarer Herzkrankheit, mit Vorsicht anwenden. Bei Patienten, bei denen Befunde oder Symptome einer akuten Myokardischämie auftreten, ist eine Unterbrechung der Behandlung in Betracht zu ziehen.
Aneurysmen und Arteriendissektionen
Die Verwendung von VEGF-Signalweg-Hemmern bei Patienten mit oder ohne Hypertonie kann die Entstehung von Aneurysmen und/oder Arteriendissektionen begünstigen. Vor Beginn der Behandlung mit Ofev sollte dieses Risiko bei Patienten mit Risikofaktoren wie Hypertonie oder Aneurysmen in der Vorgeschichte sorgfältig abgewogen werden.
Venöse Thromboembolien
In den klinischen Studien wurde bei den mit Nintedanib behandelten Patienten kein erhöhtes Risiko für venöse Thromboembolien beobachtet. Angesichts des Wirkmechanismus von Nintedanib könnte ein erhöhtes Risiko für thromboembolische Ereignisse bestehen.
Gastrointestinale Perforationen
Mit Nintedanib behandelte Patienten können aufgrund des Wirkmechanismus des Arzneimittels ein erhöhtes Risiko für gastrointestinale Perforationen aufweisen. In den INPULSIS-Studien (IPF-Patienten) wurde bei 0,3 % der mit Ofev und bei keinem (0) der mit Placebo behandelten Patienten eine gastrointestinale Perforation beschrieben. In der SENSCIS-Studie (SSc-ILD-Patienten) und der INBUILD-Studie (Patienten mit progressiven fibrosierenden ILDs) wurden in beiden Behandlungsgruppen keine Fälle von gastrointestinaler Perforation berichtet.
Nach Markteinführung wurden Fälle von gastrointestinalen Perforationen, darunter auch tödliche, berichtet. Bei Patienten mit kürzlichem bauchchirurgischem Eingriff, kürzlich aufgetretener Perforation eines Hohlorgans, peptischen Ulzera in der Anamnese, Divertikulose oder bei begleitender Anwendung von Corticosteroiden oder NSAR ist besondere Vorsicht geboten. Bei Auftreten einer gastrointestinalen Perforation ist die Behandlung mit Ofev endgültig abzusetzen.
Patienten mit bekanntem Risiko für eine gastrointestinale Perforation dürfen nur mit Ofev behandelt werden, wenn der erwartete Nutzen das potenzielle Risiko überwiegt.
Wundheilungsstörungen
In den klinischen Studien wurde keine erhöhte Inzidenz von Wundheilungsstörungen beobachtet. Nintedanib kann angesichts seines Wirkmechanismus die Wundheilung beeinträchtigen. Es wurden keine Studien durchgeführt, die eigens die Auswirkungen von Nintedanib auf die Wundheilung untersuchten. Daher sollte die Behandlung mit Ofev erst dann begonnen – bzw. bei perioperativer Unterbrechung der Behandlung wieder aufgenommen – werden, wenn nach klinischem Ermessen eine ausreichende Wundheilung erfolgt ist.
Sojalecithin
Ofev Weichkapseln enthalten Sojalecithin (siehe «Kontraindikationen»).
Interaktionen
Studien zur Erfassung von Wechselwirkungen wurden nur bei Erwachsenen durchgeführt.
Einfluss anderer Wirkstoffe auf Nintedanib:
P-Glycoprotein (P-gp) und CYP3A4 Inhibitoren und Induktoren
Nintedanib ist ein Substrat von P-gp (siehe «Pharmakokinetik») und zu einem geringen Teil auch von CYP3A4. Die gemeinsame Verabreichung mit dem starken P-gp-Inhibitor und CYP3A4 Inhibitor Ketoconazol bewirkte einen Anstieg der AUC von Nintedanib auf das 1,61-fache und der Cmax auf das 1,83-fache.
Starke P-gp-Inhibitoren (z.B. Ketoconazol oder Erythromycin) können bei gemeinsamer Verabreichung mit Ofev die Nintedanib-Exposition erhöhen. In solchen Fällen ist eine engmaschige Überwachung der Verträglichkeit von Nintedanib geboten. Nebenwirkungen können eine Unterbrechung der Behandlung mit Ofev, eine Dosisreduktion oder ein Absetzen von Ofev erforderlich machen (siehe «Dosierung/Anwendung»).
Bei gleichzeitiger Verabreichung mit dem starken P-gp- und CYP3A4 Induktor Rifampicin nahm die AUC von Nintedanib auf 50,3 % und die Cmax auf 60,3 % des bei alleiniger Verabreichung von Nintedanib gemessenen Wertes ab.
Starke P-gp-Induktoren (z.B. Rifampicin, Carbamazepin, Phenytoin und Johanniskraut) können die Nintedanib-Exposition verringern. Die Auswahl einer anderen Begleitmedikation ohne oder mit minimalem Potential für eine P-gp-Induktion ist zu erwägen.
Nahrung
Ofev soll zusammen mit Nahrung eingenommen werden um die Bioverfügbarkeit zu erhöhen (siehe «Pharmakokinetik»).
pH-Wert
Die Löslichkeit von Nintedanib ist stark vom pH-Wert abhängig und sinkt bei einem pH-Wert >3. In klinischen Studien hatte die gleichzeitige Einnahme von Medikamenten, die zu einer Erhöhung des Magen pH-Wertes führen, bei Einnahme von Nintedanib mit Nahrung keinen Einfluss auf die Ctrough Werte von Nintedanib. Bei nüchterner Einnahme könnte die gleichzeitige Einnahme von Medikamenten, die zu einer Erhöhung des Magen pH-Wertes führen, die Bioverfügbarkeit von Nintedanib reduzieren. Nintedanib sollte daher mit Nahrung eingenommen werden.
Rauchen
Rauchen steht mit einer reduzierten Nintedanib Exposition im Zusammenhang, was zu einer veränderten Wirksamkeit führen könnte. Patienten sollten angehalten werden vor Beginn einer Therapie mit Ofev mit dem Rauchen aufzuhören oder das Rauchen während der Therapie mit Ofev zu reduzieren.
In-vitro-Untersuchungen zufolge ist Nintedanib kein Substrat von OATP-1B1, OATP-1B3, OATP-2B1, OCT-2, BCRP oder MRP-2. In-vitro-Studien zeigten, dass Nintedanib ein Substrat von OCT-1 ist. Es wird davon ausgegangen, dass diese Beobachtungen geringe klinische Relevanz haben.
Einfluss von Nintedanib auf andere Wirkstoffe:
Transporter
In vitro wurde eine schwache hemmende Wirkung auf OCT-1, BCRP und P-gp beobachtet. Die klinische Relevanz wurde nicht untersucht, wird aber als gering betrachtet.
In-vitro-Untersuchungen zufolge ist Nintedanib kein Inhibitor von OATP-1B1, OATP-1B3, OATP-2B1, OCT-2, oder MRP-2.
Der Metabolit BIBF 1202 war in vitro ein schwacher Hemmer von OATP-1B1, OATP-1B3, OATP-2B1, OCT-1 und BCRP. Die klinische Relevanz wurde nicht untersucht, wird aber als gering betrachtet.
Cytochrom-(CYP-)Enzyme
Die Biotransformation von Nintedanib erfolgt nur in geringem Masse über CYP-Enzyme. Nintedanib und seine Metaboliten, die freie Säure BIBF 1202 und ihr Glucuronid BIBF-1202-Glucuronid, hatten in präklinischen Studien weder einen hemmenden, noch einen induzierenden Einfluss auf CYP-Enzyme (siehe «Pharmakokinetik»). Die Wahrscheinlichkeit für Arzneimittelwechselwirkungen aufgrund von Beeinflussung von CYP-Stoffwechselwegen durch Nintedanib wird daher als gering eingeschätzt.
Gemeinsame Verabreichung mit anderen Arzneimitteln
Begleittherapie mit Pirfenidon:
Ausgehend von den Ergebnissen einer speziellen Pharmakokinetik (PK)-Studie, gibt es keine Hinweise auf eine relevante pharmakokinetische Interaktion zwischen Nintedanib und Pirfenidon, wenn diese Wirkstoffe in Kombination verabreicht werden.
Begleittherapie mit Bosentan
In einer speziellen Pharmakokinetik (PK)-Studie wurde die gleichzeitige Behandlung von Ofev mit Bosentan an gesunden Freiwilligen untersucht. Die Studienteilnehmer erhielten eine Einzeldosis von 150 mg Ofev vor und nach einer Mehrfachgabe von 125 mg Bosentan zweimal täglich im Steady State. Die adjustierten Verhältnisse der geometrischen Mittelwerte (90% Konfidenzintervall [KI]) lagen bei 103% (86% - 124%) bzw. 99% (91% - 107%) für die Cmax bzw. AUC0-tz von Nintedanib (n=13), was darauf hinweist, dass die gleichzeitige Gabe von Nintedanib mit Bosentan die Pharmakokinetik von Nintedanib nicht veränderte.
Das Potential für Wechselwirkungen zwischen Nintedanib und hormonellen Kontrazeptiva wurde nicht untersucht.
Schwangerschaft/Stillzeit
Schwangerschaft
Es liegen keine Daten zur Anwendung von Ofev bei Schwangeren vor, allerdings zeigten präklinische tierexperimentelle Studien eine Reproduktionstoxizität des Arzneimittels (siehe «Präklinische Daten»). Da Nintedanib auch beim Menschen den Fötus schädigen kann, darf es in der Schwangerschaft nicht angewendet werden.
Vor Beginn der Behandlung mit Ofev ist ein Schwangerschaftstest vorzunehmen, der während der Behandlung nach Bedarf zu wiederholen ist.
Weibliche Patienten sind darauf hinzuweisen, dass sie ihren Arzt oder Apotheker informieren müssen, wenn sie während der Behandlung mit Ofev schwanger werden.
Wird eine Patientin während der Behandlung mit Ofev schwanger, ist die Behandlung abzusetzen und die Patientin sollte auf die potenziellen Risiken für den Fötus hingewiesen werden.
Empfängnisverhütung
Nintedanib kann den Fötus schädigen (siehe «Präklinische Daten»).
Mit Ofev behandelte Frauen, die schwanger werden können, sollten darauf hingewiesen werden, dass sie während der Behandlung mit Ofev nicht schwanger werden sollen und dass während und bis mindestens 3 Monate nach der letzten Dosis Ofev hoch wirksame Verhütungsmethoden anzuwenden sind.
Da derzeit nicht bekannt ist, ob Nintedanib die Wirksamkeit hormoneller Verhütungsmittel einschränken kann, müssen Frauen, die hormonelle Verhütungsmittel anwenden, zusätzlich eine Barrieremethode anwenden.
Stillzeit
Es liegen keine Daten zur Ausscheidung von Nintedanib und seinen Metaboliten in die Muttermilch vor.
In präklinischen Studien wurden bei säugenden Ratten geringe Mengen an Nintedanib und seinen Metaboliten (≤0,5 % der verabreichten Dosis) in der Milch nachgewiesen.
Ein Risiko für das Neugeborene / Kind kann nicht ausgeschlossen werden. Das Stillen soll während der Behandlung mit Ofev unterbrochen werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Es wurden keine Studien zu den Auswirkungen auf die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt.
Die Patienten sind darauf hinzuweisen, dass sie während einer Behandlung mit Ofev beim Führen eines Fahrzeugs und beim Bedienen von Maschinen vorsichtig sein sollten.
Unerwünschte Wirkungen
Die Sicherheit von Ofev wurde in klinischen Studien bei mehr als 1'000 Patienten mit IPF untersucht, von denen mehr als 200 Ofev mehr als 2 Jahre lang erhielten.
Ofev wurde in drei 52-wöchigen randomisierten, placebokontrollierten Doppelblind-Studien bei Patienten mit IPF untersucht. In der Phase-II-Studie (TOMORROW) und den Phase-III-Studien (INPULSIS-1 und INPULSIS-2) erhielten 723 Patienten mit IPF Ofev 150 mg zweimal täglich und 508 Patienten Placebo. Die mediane Dauer der Exposition betrug bei mit Ofev behandelten Patienten 10 Monate und bei mit Placebo behandelten Patienten 11 Monate. Die Studienteilnehmer waren zwischen 42 und 89 Jahre alt (medianes Alter von 67 Jahren). Die meisten Patienten waren Männer (79 %) und kaukasischer Abstammung (60 %).
Ofev wurde zudem in einer randomisierten, doppelblinden, placebokontrollierten Phase-III-Studie (INBUILD) untersucht, in der die Behandlung mit Ofev 150 mg zweimal täglich (n=332) mit Placebo (n=331) an 663 Patienten mit anderen chronischen fibrosierenden ILDs mit einem progressiven Phänotyp verglichen wurde. Während die Patienten mindestens 52 Wochen behandelt wurden, dauerte die Behandlung bei einzelnen Patienten bis zu 27 Monate. Die mediane Expositionsdauer betrug 16 Monate bei mit Ofev behandelten Patienten und 17 Monate bei Patienten, die Placebo erhielten. Das Alter der Studienteilnehmer reichte von 27 bis 87 Jahren (medianes Alter: 67 Jahre). Die meisten Patienten waren männlich (54 %). Die Patienten waren überwiegend Kaukasier (74 %) oder Asiaten (25 %).
Ofev wurde in einer randomisierten, doppelblinden, placebokontrollierten Phase‑III-Studie (SENSCIS) untersucht, in der 576 Patienten mit SSc-ILD zweimal täglich 150 mg Ofev (n=288) oder Placebo (n=288) erhielten. Während die Patienten mindestens 52 Wochen behandelt wurden, dauerte die Behandlung bei einzelnen Patienten bis zu 100 Wochen. Die mediane Expositionsdauer betrug 15 Monate bei mit Ofev behandelten Patienten und 16 Monate bei Patienten, die Placebo erhielten. Das Alter der Probanden reichte von 20 bis 79 Jahre (medianes Alter: 55 Jahre). Die meisten Patienten waren weiblich (75 %). Die Patienten waren überwiegend Kaukasier (67 %), Asiaten (25 %) oder hatten eine schwarze Hautfarbe (6 %). 49 % der Patienten erhielten zu Studienbeginn eine stabile Therapie mit Mycophenolat.
Die häufigsten im Zusammenhang mit der Anwendung von Ofev beschriebenen Nebenwirkungen beinhalteten Diarrhö, Übelkeit und Erbrechen, abdominelle Schmerzen, Appetitabnahme, Gewichtsabnahme, Blutungen, Erhöhung der Leberenzyme und Hautausschlag.
Das Sicherheitsprofil bei mit Ofev behandelten Patienten war vergleichbar, und zwar unabhängig davon, ob sie zu Studienbeginn Mycophenolat erhielten oder nicht.
Der Abschnitt «Warnhinweise und Vorsichtsmassnahmen» enthält Hinweise zum Vorgehen bei ausgewählten Nebenwirkungen.
Bei den Nebenwirkungshäufigkeiten werden folgende Häufigkeitskategorien zugrunde gelegt: «sehr häufig» (≥1/10), «häufig» (< 1/10, ≥1/100), «gelegentlich» (< 1/100, ≥1/1000), «selten» (< 1/1000, ≥1/10'000), «sehr selten» (< 1/10'000).
Idiopathische Lungenfibrose (IPF)
Erkrankungen des Blutes und des Lymphsystems
Gelegentlich: Thrombozytopenie.
Stoffwechsel- und Ernährungsstörungen
Häufig: Appetitverlust, Gewichtsverlust.
Erkrankungen des Nervensystems
Häufig: Kopfschmerzen.
Gefässerkrankungen
Häufig: Blutungena.
Gelegentlich: Hypertonieb.
Häufigkeit nicht bekannt: Aneurysmen und Arteriendissektionen.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Diarrhö (53,6%), Übelkeit (19,1%), abdominelle Schmerzenc (10,2%).
Häufig: Erbrechen.
Gelegentlich: Pankreatitis, gastrointestinale Perforation, Kolitis.
Affektionen der Leber und Gallenblase
Sehr häufig: Erhöhung der Leberenzyme (10,5%).
Häufig: Erhöhung der Alaninaminotransferase (ALT), Erhöhung der Aspartataminotransferase (AST), Erhöhung der Gammaglutamyltransferase (GGT).
Gelegentlich: Erhöhung der Alkalischen Phosphatase (ALP) im Blut, Hyperbilirubinämie, Arzneimittelbedingter Leberschaden.
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Hautausschlag.
Gelegentlich: Pruritus, Alopezie.
Erkrankungen der Nieren und Harnwege
Häufigkeit nicht bekannt: Nierenversagen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
a Nach Markteinführung wurden nicht schwerwiegende und schwerwiegende Blutungsereignisse, darunter tödliche, beobachtet, die mit den Erfahrungen aus klinischen Studien übereinstimmen. Die häufigsten Blutungsereignisse umfassten rektale Blutung/Hämatochezie, Nasenbluten sowie Bluterguss. Tödliche Ereignisse betrafen gastrointestinale, intrakranielle und pulmonale Blutungen.sowie DIC.
b Umfasst Hypertonie, Blutdruck erhöht, hypertensive Krise und hypertensive Kardiomyopathie.
c Umfasst Abdominalschmerz, Schmerzen Oberbauch, Schmerzen Unterbauch, gastrointestinale Schmerzen, abdominaler Druckschmerz.
Andere chronische fibrotisierende interstitielle Lungenerkrankungen (ILD) mit einem progressiven Phänotyp
Erkrankungen des Blutes und des Lymphsystems
Gelegentlich: Thrombozytopenie.
Stoffwechsel- und Ernährungsstörungen
Sehr häufig: Appetitverlust (11,1%).
Häufig: Gewichtsverlust.
Erkrankungen des Nervensystems
Häufig: Kopfschmerzen.
Gefässerkrankungen
Häufig: Hypertonie, Blutungen.
Häufigkeit nicht bekannt: Aneurysmen und Arteriendissektionen.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Diarrhö (59%), Übelkeit (24%), abdominelle Schmerzen (11%), Erbrechen (12%).
Gelegentlich: Pankreatitis, Kolitis.
Häufigkeit nicht bekannt: gastrointestinale Perforation
Affektionen der Leber und Gallenblase
Sehr häufig: Erhöhung der Leberenzyme (18,4%), Erhöhung der Alaninaminotransferase (ALT) (10,8%).
Häufig: Erhöhung der Aspartataminotransferase (AST), Erhöhung der Gammaglutamyltransferase (GGT), Erhöhung der Alkalischen Phosphatase (ALP) im Blut, Arzneimittelbedingter Leberschaden.
Gelegentlich: Hyperbilirubinämie.
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Hautausschlag.
Gelegentlich: Pruritus, Alopezie.
Erkrankungen der Nieren und Harnwege
Häufigkeit nicht bekannt: Nierenversagen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Mit systemischer Sklerose assoziierte interstitielle Lungenerkrankung (SSc-ILD)
Erkrankungen des Blutes und des Lymphsystems
Gelegentlich: Thrombozytopenie.
Stoffwechsel- und Ernährungsstörungen
Häufig: Appetitverlust, Gewichtsverlust.
Erkrankungen des Nervensystems
Häufig: Kopfschmerzen.
Gefässerkrankungen
Häufig: Hypertonie, Blutungen.
Häufigkeit nicht bekannt: Aneurysmen und Arteriendissektionen.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Diarrhö (68,4%), Übelkeit (24,7%), abdominelle Schmerzen (11,8%), Erbrechen (17,7%).
Gelegentlich: Kolitis.
Häufigkeit nicht bekannt: Pankreatitis, gastrointestinale Perforation.
Affektionen der Leber und Gallenblase
Sehr häufig: Erhöhung der Leberenzyme (11,1%).
Häufig: Erhöhung der Alaninaminotransferase (ALT), Erhöhung der Aspartataminotransferase (AST), Erhöhung der Gammaglutamyltransferase (GGT), Erhöhung der Alkalischen Phosphatase (ALP) im Blut.
Gelegentlich: Arzneimittelbedingter Leberschaden.
Häufigkeit nicht bekannt: Hyperbilirubinämie.
Erkrankungen der Haut und des Unterhautzellgewebes
Gelegentlich: Hautausschlag, Pruritus.
Häufigkeit nicht bekannt: Alopezie.
Erkrankungen der Nieren und Harnwege
Gelegentlich: Nierenversagen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Es gibt kein spezifisches Antidot und keine spezifische Behandlung für eine Überdosierung mit Ofev. Die höchste in Phase-I-Studien verabreichte Nintedanib-Einzeldosis betrug 450 mg einmal täglich. Darüber hinaus erhielten 2 Patienten bis zu acht Tage lang eine Überdosierung mit maximal 600 mg zweimal täglich. Die beobachteten unerwünschten Ereignisse standen im Einklang mit dem bekannten Sicherheitsprofil von Nintedanib, d.h. mit erhöhten Leberenzymen und gastrointestinalen Symptomen. Beide Patienten erholten sich von diesen Nebenwirkungen.
In den INPULSIS-Studien (IPF-Patienten) erhielt ein Patient versehentlich insgesamt 21 Tage lang eine Dosis von 600 mg pro Tag. In der Phase der fehlerhaften Dosierung trat ein nicht-schwerwiegendes unerwünschtes Ereignis (Nasopharyngitis) auf und klang wieder ab. Es traten keine weiteren beschriebenen Ereignisse auf.
Im Fall einer Überdosierung sollte die Behandlung unterbrochen werden und es sollten geeignete allgemeine supportive Massnahmen eingeleitet werden.
Eigenschaften/Wirkungen
ATC-Code
L01XE31
Wirkungsmechanismus
Nintedanib ist ein Tyrosinkinaseinhibitor vom Typ der kleinen Moleküle. Zu den inhibierten Tyrosinkinasen gehören die Rezeptoren Platelet-Derived Growth Factor-Receptor (PDGFR) α und β, Fibroblastenwachstumsfaktorrezeptor (FGFR) 1-3 und Vaskulärer Endothelialer Wachstumsfaktorrezeptor (VEGFR) 1-3. Nintedanib bindet kompetitiv an die ATP-Bindungstasche dieser Rezeptoren und blockiert die intrazelluläre Signalgebung, die für die Proliferation, Migration und Transformation von Fibroblasten – die essentielle Pathomechanismen der IPF sind – entscheiden ist. Darüber hinaus hemmt Nintedanib die Kinasen Flt-3, Lck, Lyn und Src.
Pharmakodynamik
Die Aktivierung der FGFR- und PDGFR-Signalkaskaden ist ein entscheidender Faktor für die Proliferation und Migration von Lungenfibroblasten/Myofibroblasten, den kennzeichnenden Zellen der Pathologie der idiopathischen Lungenfibrose. Die potenzielle Bedeutung der Hemmung von VEGFR für die Pathologie der IPF wurde noch nicht vollständig aufgeklärt. Es wird angenommen, dass Nintedanib auf molekularer Ebene die FGFR- und PDGFR-Signalkaskaden hemmt, die die Proliferation und Migration von Lungenfibroblasten vermitteln, indem es an die Adenosintriphosphat-(ATP)Bindungstasche der intrazellulären Rezeptorkinasedomäne bindet und auf diese Weise die Kreuzaktivierung (über Autophosphorylierung der Rezeptor-Homodimere) beeinträchtigt.
In vitro hemmt Nintedanib seine Zielrezeptoren in Konzentrationen im niedrigen nanomolaren Bereich. An Lungenfibroblasten von Patienten mit IPF hemmte Nintedanib die durch PDGF, FGF und VEGF stimulierte Proliferation mit EC50-Werten von 11 nmol/l, 5,5 nmol/l bzw. unter 1 nmol/l. In Konzentrationen zwischen 100 und 1'000 nmol/l hemmte Nintedanib darüber hinaus die durch PDGF, FGF und VEGF stimulierte Fibroblastenmigration und die TGF-β2-induzierte Transformation von Fibroblasten in Myofibroblasten. Darüber hinaus wird angenommen, dass die antiinflammatorische Aktivität von Nintedanib die fibrotische Stimulation begrenzt, indem sie profibrotische Mediatoren wie IL-1β und IL-6 reduziert. Es wurde noch nicht geklärt, welchen Anteil die antiangiogene Aktivität von Nintedanib am Wirkmechanismus des Arzneimittels bei fibrotischen Lungenerkrankungen hat. In In-vivo-Studien hatte Nintedanib eine starke antifibrotische und antiinflammatorische Aktivität.
Klinische Wirksamkeit
Idiopathische Lungenfibrose (IPF)
Die klinische Wirksamkeit von Nintedanib wurde in einer Phase-II-Studie (TOMORROW) und zwei Phase-III-Studien (INPULSIS-1 und INPULSIS-2) bei 1231 Patienten mit IPF untersucht. Bei den genannten Studien handelte es sich um randomisierte, doppelblinde, placebokontrollierte Studien, in denen eine 52-wöchige Behandlung mit Ofev 150 mg zweimal täglich mit Placebo verglichen wurde.
Die Studien INPULSIS-1 und INPULSIS-2 hatten ein identisches Studiendesign und das Design der TOMORROW-Studie war sehr ähnlich. Die Patienten wurden in einem Verhältnis von 3:2 (in der TOMORROW-Studie 1:1) randomisiert einer 52-wöchigen Behandlung mit Ofev 150 mg oder Placebo, jeweils zweimal täglich, zugeteilt. Die TOMORROW-Studie besass darüber hinaus weitere Behandlungsarme (50 mg pro Tag, 50 mg zweimal täglich und 100 mg zweimal täglich), die hier nicht weiter besprochen werden.
Primärer Endpunkt war die jährliche Abnahme der forcierten Vitalkapazität (FVC). Die Änderung des Gesamtscores im Saint George's Respiratory Questionnaire (SGRQ) zwischen Studienbeginn und Woche 52 und die Zeit bis zur ersten akuten IPF-Exazerbation waren wichtige sekundäre Endpunkte der INPULSIS-Studien und sekundäre Endpunkte der TOMORROW-Studie.
Jährliche Abnahme der FVC
Die jährliche Abnahme der FVC (in ml) war bei mit Nintedanib behandelten Patienten signifikant geringer ausgeprägt als bei mit Placebo behandelten Patienten. Die Wirkung der Behandlung war in allen 3 Studien einheitlich. Tabelle 1 enthält die Ergebnisse der Einzelstudien und der gepoolten INPULSIS-Analyse.
Tabelle 1 Jährliche Abnahme der FVC (ml) in den Studien TOMORROW, INPULSIS-1 und INPULSIS-2 sowie der gepoolten INPULSIS-Analyse – behandelte Population2
TOMORROW | INPULSIS-1 | INPULSIS-2 | INPULSIS-1 und INPULSIS-2 gepoolt | |||||
|---|---|---|---|---|---|---|---|---|
Placebo | Ofev 150 mg zweimal täglich | Placebo | Ofev 150 mg zweimal täglich | Placebo | Ofev 150 mg zweimal täglich | Placebo | Ofev 150 mg zweimal täglich | |
Anzahl der ausgewerteten Patienten | 83 | 84 | 204 | 309 | 219 | 329 | 423 | 638 |
Rate 1,2 (SE) der Abnahme über 52 Wochen | -190 (36) | -60 (39) | −239,9 (18,71) | −114,7 (15,33) | −207,3 (19,31) | −113,6 (15,73) | −223,5 (13,45) | −113,6(10,98) |
Vergleich vs. Placebo | ||||||||
Differenz1 | 131 | 125,3 | 93,7 | 109,9 | ||||
95%-KI | (27; 235) | (77,7; 172,8) | (44,8; 142,7) | (75,9; 144,0) | ||||
p-Wert | 0,01363 | <0,0001 | 0,0002 | <0,0001 | ||||
1 Berechnung auf Grundlage eines Regressionsmodells mit Zufallskoeffizienten. 2 Randomisierte Population in der TOMORROW-Studie; behandelte Population in INPULSIS-1, INPULSIS-2 und den gepoolten INPULSIS-Daten 3 Nominaler p-Wert. | ||||||||
Alle präspezifizierten Sensitivitätsanalysen bestätigten die Robustheit der Wirkung von Nintedanib im Sinne einer Reduktion der jährlichen Abnahme der FVC. Darüber hinaus wurden vergleichbare Auswirkungen auf andere Endpunkte der Lungenfunktion wie beispielsweise die Änderung der FVC in Woche 52 gegenüber dem Ausgangwert und die FVC-Responder-Analyse beobachtet, was die Wirkungen von Nintedanib im Sinne einer Verlangsamung des Fortschreitens der Erkrankung untermauert. Abbildung 1 zeigt den Verlauf der Änderung gegenüber dem Ausgangswert über die Zeit in beiden Behandlungsgruppen (auf Grundlage der gepoolten Analyse der Studien INPULSIS-1 und INPULSIS-2).
Abbildung 1: Mittlere (SEM) beobachtete Änderung der FVC gegenüber dem Ausgangswert (ml) über die Zeit, Studien INPULSIS-1 und INPULSIS-2 (gepoolte Daten)

bid = zweimal täglich, SEM = Standardfehler des Mittelwerts
Änderung im SGRQ-Gesamtscore in Woche 52 gegenüber dem Ausgangswert
Der Gesamtscore im St. George's Respiratory Questionnaire (SGRQ) ist ein Mass für die gesundheitsbezogene Lebensqualität (HRQoL) und wurde in Woche 52 ausgewertet. In der TOMORROW-Studie betrug die geschätzte mittlere Änderung im SGRQ-Gesamtscore zwischen Studienbeginn (Baseline) und Woche 52 unter Placebo 5,46, was auf eine Verschlechterung der gesundheitsbezogenen Lebensqualität hinweist, und unter Nintedanib -0,66, was eine stabile gesundheitsbezogene Lebensqualität anzeigt. Die geschätzte mittlere Differenz zwischen Nintedanib und Placebo betrug -6,12 (95%-KI: -10,57; -1,67; p = 0,0071).
In der Studie INPULSIS-2 wiesen die mit Placebo behandelten Patienten einen ausgeprägteren Anstieg des SGRQ-Gesamtscores gegenüber dem Ausgangswert auf als die mit Nintedanib 150 mg zweimal täglich behandelten Patienten. Die Verschlechterung der HRQoL war in der Nintedanib-Gruppe geringer ausgeprägt. Die Differenz zwischen den Behandlungsgruppen war statistisch signifikant (-2,69; 95%-KI: -4,95; -0,43; p = 0,0197).
In der Studie INPULSIS-1 fiel der Anstieg des SGRQ-Gesamtscores in Woche 52 gegenüber dem Ausgangswert in der Nintedanib- und Placebogruppe vergleichbar aus (Differenz zwischen den Behandlungsgruppen: -0,05; 95%-KI: -2,50; 2,40; p = 0,9657). In der gepoolten Analyse der INPULSIS-Studien war die geschätzte mittlere Änderung im SGRQ-Gesamtscore zwischen Ausgangswert und Woche 52 in der Nintedanib-Gruppe (3,53) geringer als in der Placebogruppe (4,96). Die Differenz zwischen den Behandlungen betrug -1,43 (95%-KI: -3,09; 0,23; p = 0,0923). Insgesamt ist der über den SGRQ-Gesamtscore beurteilte Einfluss von Nintedanib auf die gesundheitsbezogene Lebensqualität moderat und weist auf eine geringer ausgeprägte Verschlechterung hin als unter Placebo.
Zeit bis zur ersten akuten IPF-Exazerbation
In den Studien TOMORROW und INPULSIS-2 war das Risiko für eine erste akute IPF-Exazerbation über den Zeitraum von 52 Wochen bei mit Nintedanib behandelten Patienten signifikant geringer als bei mit Placebo behandelten Patienten (Hazard Ratio [HR]: 0,16; 95%-KI: 0,04; 0,71; p = 0,0054) bzw. (HR: 0,38; 95%-KI: 0,19; 0,77; p = 0,005). In der Studie INPULSIS-1 gab es keine Differenz zwischen den Behandlungsgruppen (HR: 1,15; 95%-KI: 0,54; 2,42; p = 0,6728).
Überlebensanalyse
In der präspezifizierten gepoolten Analyse der Überlebensdaten der INPULSIS-Studien fiel die Gesamtmortalität über 52 Wochen in der Nintedanib-Gruppe (5,5 %) numerisch niedriger aus als in der Placebogruppe (7,8 %). Die Analyse der Zeit bis zum Tod ergab eine HR von 0,70 (95%-KI: 0,43; 1,12; p = 0,1399). Die Ergebnisse für alle Überlebensendpunkte (wie Mortalität während der Behandlung und respiratorische Mortalität) zeigten einheitlich numerische Differenzen zugunsten von Nintedanib, die jedoch keine statistische Signifikanz erreichten.
Weitere Daten aus der Phase-IV-Studie INJOURNEY mit zweimal täglicher Gabe von 150 mg Ofev und zusätzlich Pirfenidon:
In einer exploratorischen, unverblindeten, randomisierten Studie wurde die gemeinsame Gabe von 150 mg Nintedanib zweimal täglich plus Pirfenidon (Titration auf 801 mg dreimal täglich) im Vergleich zur alleinigen Gabe von 150 mg Nintedanib zweimal täglich bei 105 Patienten über 12 Wochen untersucht. Primärer Endpunkt war der Prozentsatz an Patienten mit unerwünschten gastrointestinalen Ereignissen von Studienbeginn bis Woche 12. Gastrointestinale unerwünschte Ereignisse traten häufig auf, was mit dem belegten Sicherheitsprofil für jede der beiden Substanzen übereinstimmt. Diarrhoe, Übelkeit und Erbrechen waren die häufigsten unerwünschten Ereignisse, die bei 20 (37,7%) gegenüber 16 (31,4%), 22 (41,5%) gegenüber 6 (11,8%) bzw. 15 (28,3%) gegenüber 6 (11,8%) der Patienten nach Behandlung mit Pirfenidon zusätzlich zu Nintedanib gegenüber Nintedanib allein gemeldet wurden.
Die mittleren (SE) absoluten Änderungen der FVC gegenüber den Ausgangswerte nach 12 Wochen waren −13,3 (17,4) mL in Patienten, welche mit Pirfenidon zusätzlich zu Nintedanib behandelt wurden (n=48) gegenüber −40,9 (31,4) mL in Patienten, welche mit Nintedanib allein behandelt wurden (n=44).
Chronisch fibrosierende Interstitielle Lungenerkrankungen (ILD) mit einem progressiven Phänotyp
Die klinische Wirksamkeit von OFEV bei Patienten mit chronisch fibrosierender ILD mit einem progressiven Phänotyp wurde in einer randomisierten, doppelblinden, Placebo-kontrollierten Phase 3 Studie (INBUILD) untersucht.
Insgesamt wurden n=663 Patienten 1:1 randomisiert. Sie erhielten entweder OFEV 2x150mg pro Tag oder Placebo für mindestens 52 Wochen.
Die Randomisierung wurde stratifiziert gemäss Fibrosemuster in der hochauflösenden Computertomographie (HRCT). N=412 Patienten mit einem UIP (usual interstitial pneumonia)-artigen Fibrosemuster und n=251 Patienten mit anderen Fibrosemustern wurden randomisiert. Für die Studienauswertung wurden 2 co-primäre Populationen definiert: die Gesamtpopulation und die Population mit UIP-artigem Fibrosemuster gemäss HRCT.
Der primäre Studienendpunkt war die jährliche Abnahme der FVC (ml) über 52 Wochen. Weitere Studienendpunkte waren beispielsweise die Zeit bis zur ersten akuten ILD Exazerbation oder zum Tod oder die Zeit bis zum Tod.
Patienten mit der Diagnose einer chronisch fibrosierenden ILD wurden für die Studie berücksichtigt, wenn sie im HRCT eine relevante Fibrosierung aufwiesen (>10% Fibrosierung) und klinische Merkmale einer Krankheitsprogression (definiert als Abnahme der FVC ≥10%, Abnahme der FVC zwischen 5 und 10% mit Verschlechterung in Krankheitssymptomen ODER Bildgebung oder Verschlechterung in Krankheitssymptomen UND Bildgebung) über die vorausgehenden 24 Monate trotz adäquater Behandlung aufwiesen. Die FVC musste ≥45% predicted und die DLCO zwischen 30% und 80% predicted betragen.
Von einer Teilnahme an der Studie ausgeschlossen waren Patienten mit
- IPF, relevanter Bronchokonstriktion (z.B. FEV1/FVC <0,7 vor Bronchodilatation) oder signifikanter pulmonaler Hypertonie
- Erhöhungen der Transaminasen oder des Bilirubins >1,5 ULN
- bekanntem Blutungsrisiko, voller Antikoagulation, kürzlichem Myokardinfarkt oder Schlaganfall
- vorausgegangener Behandlung mit Nintedanib, Pirfenidon, Azathioprin, Cyclosporin, Mycophenolat, Tacrolimus, oralen Kortikoiden >20mg/Tag oder oralen Kortikoiden+Azathioprin+N-Acetylcystein innert 4 Wochen vor Randomisierung*
- vorausgegangener Behandlung mit Cyclophosphamid innert 8 Wochen vor Randomisierung*
- vorausgegangener Behandlung mit Rituximab innert 6 Monaten vor Randomisierung*
* der Einsatz der genannten Medikamente war ab 6 Monaten nach Studienbeginn wieder gestattet, soweit klinisch indiziert
Die Studienteilnehmer waren im Wesentlichen Kaukasier (74%) und Asiaten (25%). 54% der Patienten waren männlich, 49% hatten niemals geraucht, das mittlere Alter betrug 66 Jahre und die FVC betrug im Mittel 69% predicted. Die zugrundeliegenden ILDs waren Hypersensitivitätspneumonien (26%), autoimmun-assoziierte ILDs (26%), idiopathische nicht-spezifische interstitielle Pneumonie (19%), unklassifizierbare idiopathische interstitielle Pneumonie (17%) und andere ILDs (12%).
Jährliche Abnahme der FVC (ml)
Über 52 Wochen wurde eine statistisch signifikante Reduktion in der Abnahme der FVC unter der Behandlung mit OFEV versus Placebo beobachtet. Der Unterschied von OFEV zu Placebo betrug 107ml (Tabelle 2).
Tabelle 2: Jährliche Abnahme der FVC (ml, primärer Studienendpunkt INBUILD Studie)
Gesamtpopulation | Subpopulation UIP-artig | Subpopulation Andere Fibrosemuster in der HRCT | ||||
|---|---|---|---|---|---|---|
OFEV | Placebo | OFEV | Placebo | OFEV | Placebo | |
Anzahl der ausgewerteten Patienten | 332 | 331 | 206 | 206 | 126 | 125 |
Ratea (SE) der Abnahme über 52 Wochen | -80,8 | -187,8 | -82,9 | -211,1 | -79,0 | -154,2 |
Vergleich vs Placebo | 107,0 | 128,2 | 75,2* | |||
95%-KI | (65,4; 148.5) | (70,8; 185,6) | (15,5; 135,0)* | |||
p-Wert | < 0,0001 | < 0,0001 | ||||
* Ein auf der Subpopulation mit anderen Fibrosemustern in der HRCT beruhender Vergleich wurde nicht in die multiple Testprozedur aufgenommen. Die hier gezeigten Werte dienen beschreibenden Zwecken. a Basierend auf einem Regressionsmodell mit zufälligen Koeffizienten und den festen kategorialen Effekten von Behandlung, HRCT-Muster, den festen kontinuierlichen Effekten von Zeit, FVC-Ausgangswert (ml) und einschliesslich der Interaktion zwischen Therapie und Zeit und der Interaktion zwischen Ausgangswert und Zeit. | ||||||
Abbildung 2 zeigt die zeitliche Entwicklung der FVC-Veränderung gegenüber Studienbeginn in den Behandlungsgruppen.
Abbildung 2. Mittlere (SEM) beobachtete Veränderung der FVC (ml) gegenüber Studienbeginn über 52 Wochen
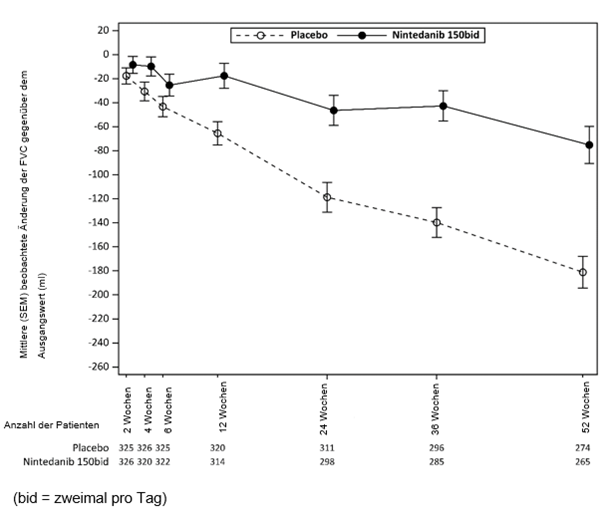
Eine post-hoc durchgeführte exploratorische Analyse nach ILD-Diagnose wurde durchgeführt (Abbildung 3). Insgesamt wurde ein konsistenter Behandlungseffekt in den unterschiedlichen ILD-Diagnosen gezeigt.
Abbildung 3: Jährliche Rate des FVC-Rückgangs (ml) über 52 Wochen basierend auf ILD-Grunddiagnose in Studie 5*
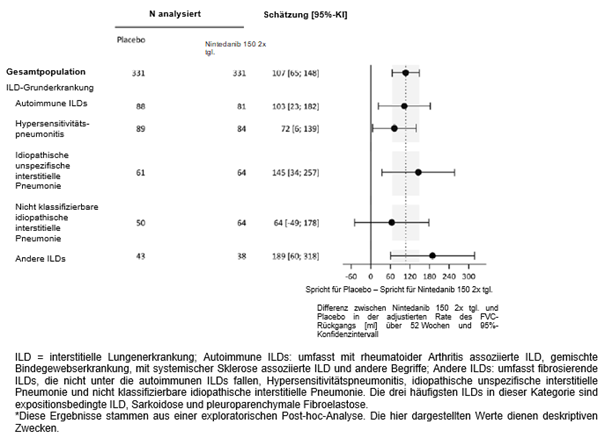
Prozentuale Abnahme der FVC
Abbildung 4 zeigt die prozentuale Veränderung der FVC (ml) von Baseline bis Woche 52. Für die Mehrzahl der Patienten fiel die Abnahme der Lungenfunktion unter OFEV geringer aus als unter Placebo.
Abbildung 4: Histogramm der prozentualen Veränderung der FVC (ml) von Baseline bis Woche 52 nach Behandlung mit prozentualen Inkrementen oder Dekrementen in 5er-Schritten (Studie 5)a
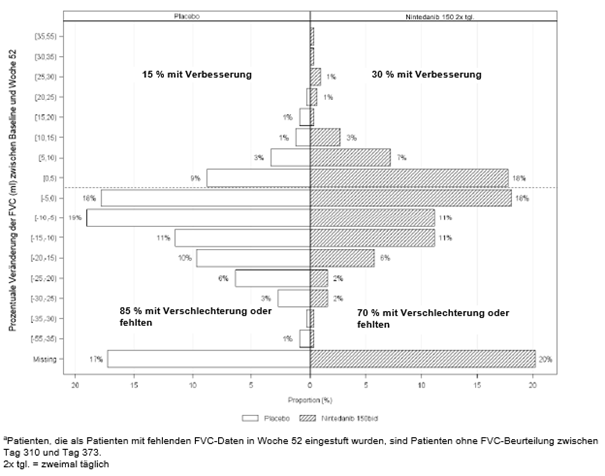
Zeit bis zur ersten akuten ILD-Exazerbation oder zum Tod
Das Risiko einer ersten akuten ILD-Exazerbation oder des Todes verminderte sich in der Ofev-Gruppe gegenüber der Placebogruppe über 52 Wochen (HR Ofev versus Placebo: 0,80; 95%-KI: 0,48; 1,34) oder über die gesamte Studiendauer (HR Ofev versus Placebo: 0,67; 95%-KI: 0,46; 0,98), was einer Reduktion des Risikos für eine erste akute ILD-Exazerbation oder den Tod um 33 % bei Patienten unter Ofev im Vergleich zu Placebo entspricht.
Überlebensanalyse
In der INBUILD-Studie wurden die Überlebensdaten unter Ofev im Vergleich zu Placebo ausgewertet, um den primären Endpunkt (FVC) zu untermauern. Die Mortalität jeglicher Ursache wurde über die gesamte Studiendauer und den verfügbaren Nachbeobachtungszeitraum beurteilt, unabhängig von der Todesursache und davon, ob die Behandlung weitergeführt wurde oder nicht. Die Analyse ergab keinen statistisch signifikanten Unterschied über 52 Wochen (HR OFEV versus Placebo 0,94: 95%-KI 0,47; 1,86) bzw. über die gesamte Studiendauer (HR OFEV versus Placebo 0,78; 95%-KI 0,5; 1,21).
Mit systemischer Sklerose assoziierte interstitielle Lungenerkrankung (SSc-ILD)
Die klinische Wirksamkeit von Nintedanib wurde in einer randomisierten, doppelblinden, placebokontrollierten Phase-III-Studie (SENSCIS) bei Patienten mit SSc-ILD untersucht.
Insgesamt wurden 580 Patienten in einem Verhältnis von 1:1 randomisiert um mindestens 52Wochen entweder Ofev 150 mg zweimal täglich oder Placebo zu erhalten. Die Randomisierung wurde nach Anti-Topoisomerase-Antikörper-(ATA)-Status stratifiziert. Einzelne Patienten setzten die verblindete Studienbehandlung bis zu 100 Wochen lang fort.
Die Diagnose der Patienten mit SSc-ILD beruhte auf den Klassifikationskriterien für SSc des American College of Rheumatology / der European League Against Rheumatism, wenn das Auftreten der Erkrankung (erstes Nicht-Raynaud-Symptom) weniger als 7 Jahre zurücklag und eine mindestens 10 %ige Lungenfibrose vorlag, ermittelt anhand eines in den vorausgegangenen 12 Monaten durchgeführten hochauflösenden Computertomografie (HRCT)-Scans.
Die FVC musste bei den Patienten mindestens 40 % des geschätzten Wertes betragen, während die DLCO 30‑89 % des geschätzten Wertes betragen musste. Patienten mit relevanten Atemwegsobstruktionen (d.h. FEV1/FVC vor Bronchodilatation unter 0,7) oder einer vorherigen oder anstehenden hämatopoetischen Stammzelltransplantation durften nicht an der Studie teilnehmen. Weitere Ausschlusskriterien waren erhöhte ALT-, AST- oder Bilirubin-Werte (>1,5xOGN), ein erhöhtes Risiko für Blutungen (einschliesslich therapeutische Antikoagulation), kürzlich erfolgter Myokardinfarkt oder Schlaganfall, eine signifikante pulmonale Hypertonie, Geschwüre an mindestens drei Fingerkuppen, eine schwere Fingernekrose mit erforderlicher Hospitalisierung in der Anamnese sowie eine sklerodermale renale Krise in der Anamnese. Patienten wurden ebenfalls ausgeschlossen, wenn sie andere Prüfmedikamente erhielten, bereits mit Nintedanib oder Pirfenidon behandelt worden waren und 8 Wochen vor Randomisierung Azathioprin oder 6 Monate vor Randomisierung Cyclophosphamid oder Cyclosporin A erhalten hatten. Eine stabile Therapie mit Mycophenolat oder Methotrexat sowie Prednison ≤10 mg/Tag oder Äquivalent war zulässig.
Primärer Endpunkt war die jährliche Abnahme der forcierten Vitalkapazität (FVC) über 52 Wochen.
Wichtige sekundäre Endpunkte waren die absolute Veränderung des modifizierten Rodnan Skin Score (mRSS) in Woche 52 gegenüber dem Ausgangswert, die absolute Veränderung des Gesamtscores im Saint George's Respiratory Questionnaire (SGRQ) in Woche 52 gegenüber dem Ausgangswert sowie die Mortalität während der gesamten Studie.
75,2 % der Patienten waren weiblich. Die Patienten waren überwiegend Kaukasier (67 %), Asiaten (25 %) oder hatten eine schwarze Hautfarbe (6 %). Das mittlere (Standardabweichung [SD], Min.-Max.) Alter betrug 54,0 (12,2, 20–79) Jahre. Insgesamt litten 51,9 % der Patienten an einer diffusen kutanen systemischen Sklerose (SSc) und 48,1 % an einer limitierten kutanen SSc. Die mittlere (SD) Zeit seit dem ersten Auftreten eines Nicht-Raynaud-Symptoms betrug 3,49 (1,7) Jahre. 49,0 % der Patienten erhielten zu Studienbeginn eine stabile Therapie mit Mycophenolat und das bei Patienten mit bzw. ohne Mycophenolat beobachtete Sicherheitsprofil war vergleichbar.
Jährliche Abnahme der FVC
Die jährliche Abnahme der FVC (in ml) über 52 Wochen wurde bei mit Ofev behandelten Patienten um 41,0 ml im Vergleich zu Placebo signifikant reduziert (siehe Tabelle 3), was einem relativen Behandlungseffekt von 43,8% entspricht.
Tabelle 3: Jährliche Abnahme der FVC (ml) über 52 Wochen
Placebo | Ofev 150 mg zweimal täglich | |
|---|---|---|
Anzahl der ausgewerteten Patienten | 288 | 287 |
Rate1 (SE) der Abnahme über 52 Wochen | -93,3 (13,5) | -52,4 (13,8) |
Vergleich vs. Placebo | ||
Differenz1 | 41,0 | |
95%-KI | (2,9, 79,0) | |
p-Wert | <0,05 | |
1 Basierend auf einer Regression mit zufälligen Koeffizienten, adjustiert für Behandlung, Geschlecht, Grösse, Alter, ATA status, FVC-Ausgangswert, FVC*Zeit und Behandlung*Zeit | ||
Eine explorative Analyse der Daten bis zu 100 Wochen (maximale Behandlungsdauer in der SENSCIS-Studie) wies darauf hin, dass die Wirkung unter einer Behandlung mit Ofev auf die Verlangsamung der Progression der SSc-ILD über 52 Wochen hinaus anhielt.
In zwei vorgegebenen Subgruppen-Wirksamkeitsanalysen wurde der mittlere Behandlungsunterschied in Bezug auf die Abnahme der FVC in Woche 52 bei den Patienten nach Region und Anwendung von Mycophenolat untersucht (siehe Abbildung 5).
Abbildung 5 Subgruppenanalyse des mittleren Behandlungsunterschiedes in Bezug auf die Abnahme der FVC (ml) in Woche 52 nach Region und Anwendung von Mycophenolat (SENSCIS)

Prozentuale Änderung der forcierten Vitalkapazität ab Studienbeginn
In Abbildung 6 wird für die SENSCIS Studie die prozentuale Änderung der FVC in ml in Woche 52 gegenüber dem Ausgangswert gezeigt. Die Verschlechterung der Lungenfunktion war bei der Mehrheit der Patienten unter Ofev geringer ausgeprägt als unter Placebo.
Abbildung 6: Histogramm der prozentualen Änderung der FVC (ml) in Woche 52 gegenüber dem Ausgangswert je nach Behandlung und prozentualer Zunahme oder Abnahme um 5 (SENSCIS)a

Abbildung 7: Mittlere (SEM) beobachtete Änderung der FVC gegenüber dem Ausgangswert (ml) über 52 Wochen

Tabelle 4: Jährliche Abnahme der FVC (% des Sollwerts) über 52 Wochen
Placebo | Ofev 150 mg zweimal täglich | |
|---|---|---|
Anzahl der ausgewerteten Patienten | 288 | 287 |
Rate1 (SE) der Abnahme über 52 Wochen | -2,6 (0,4) | -1,4 (0,4) |
Vergleich vs. Placebo | ||
Differenz1 | 1,15 | |
95%-KI | (0,09, 2,21) | |
p-Wert | <0,05 | |
1 Basierend auf einer Regression mit zufälligen Koeffizienten und den festen kategorialen Effekten von Behandlung, ATA-Status, den festen kontinuierlichen Effekten von Zeit, FVC-Ausgangswert [% des Sollwerts] und einschliesslich der Interaktion zwischen Therapie und Zeit und der Interaktion zwischen Ausgangswert und Zeit. Als zufälliger Effekt wurden patientenspezifisch Intercept und Zeit angegeben. Die Messfehler-Varianz (within-patient error) innerhalb eines Patienten wurde durch eine unstrukturierte Varianz-Kovarianz-Matrix modelliert. Die interindividuelle Variabilität wurde durch eine Varianz-Kovarianz-Matrix der Varianzkomponenten modelliert. | ||
Änderung des modifizierten Rodnan Skin Scores (mRSS) in Woche 52 gegenüber dem Ausgangswert
Die adjustierte mittlere absolute Veränderung des mRSS in Woche 52 gegenüber dem Ausgangswert war zwischen der Ofev-Gruppe (-2,17 (95%-KI -2,69, -1,65)) und der Placebogruppe (-1,96 (95%-KI -2,48, -1,45)) vergleichbar. Die adjustierte mittlere Differenz zwischen den Behandlungsgruppen betrug -0,21 (95%-KI -0,94, 0,53; p = 0,5785).
Änderung des Gesamtscores im St. George's Respiratory Questionnaire (SGRQ) in Woche 52 gegenüber dem Ausgangswert
Die adjustierte mittlere absolute Veränderung des SGRQ-Gesamtscores in Woche 52 gegenüber dem Ausgangswert war zwischen der Ofev-Gruppe (0,81 (95%-KI -0,92, 2,55)) und der Placebogruppe (-0,88 (95%-KI -2,58, 0,82)) vergleichbar. Die adjustierte mittlere Differenz zwischen den Behandlungsgruppen betrug 1,69 (95%-KI -0,73, 4,12; p = 0,1711).
Überlebensanalyse
Über die gesamte Studie war die Mortalität zwischen der Ofev-Gruppe (N = 10; 3,5 %) und der Placebogruppe (N = 9; 3,1 %) vergleichbar. Über die gesamte Studie ergab die Analyse der Zeit bis zum Tod eine HR von 1,16 (95%-KI 0,47, 2,84; p = 0,7535).
Wirkung auf das QT-Intervall
In einer speziellen Studie zu Patienten mit Nierenzellkarzinom zeigten QT/QTc-Messungen, dass eine orale Nintedanib Einzeldosis zu 200 mg und mehrmalige orale Nintedanib-Dosen zu 200 mg zweimal täglich für 15 Tage keine Verlängerung des QTcF-Intervalls zur Folge hatten. Eine vollständige Thorough-QT-Study wurde mit Nintedanib nicht durchgeführt.
Studien mit Kindern und Jugendlichen
Es wurden keine klinischen Studien mit Kindern und Jugendlichen durchgeführt.
Pharmakokinetik
Die pharmakokinetischen Eigenschaften von Nintedanib waren bei gesunden Probanden, Patienten mit IPF, SSc-ILD, ILDs und Patienten mit Krebserkrankung vergleichbar.
Die Pharmakokinetik von Nintedanib ist über die Zeit linear. Es wurde eine Dosisproportionalität mit steigender Nintedanib-Exposition bei steigenden Dosen nachgewiesen (Dosisspanne von 50 bis 450 mg einmal täglich und 150 bis 300 mg zweimal täglich). Die Akkumulation betrug nach mehrmaliger Verabreichung bei IPF Patienten in Bezug auf AUCτ das 1,76-fache. Steady-State-Plasmakonzentrationen wurden nach 1-wöchiger Verabreichung erreicht. Die Nintedanib-Talspiegel blieben über mehr als ein Jahr stabil. Die Pharmakokinetik von Nintedanib hat eine moderate bis hohe interindividuelle Variabilität (Variationskoeffizient für pharmakokinetische Standardparameter im Bereich von 30 % bis 70 %). Die intraindividuelle Variabilität ist niedrig bis moderat (Variationskoeffizient unter 40 %).
Absorption
Maximale Nintedanib-Plasmakonzentrationen wurden nach oraler Einnahme von Weichgelatinekapseln zusammen mit einer Mahlzeit innerhalb von etwa 2 4 Stunden erreicht (Spanne: 0,5 8 h). Die absolute Bioverfügbarkeit einer 100-mg-Dosis betrug bei gesunden Probanden 4,69 % (90%-KI: 3,62-6,08). Resorption und Bioverfügbarkeit werden durch den Einfluss von Transportern (P-gp), und einen substantiellen First-Pass-Metabolismus und evtl. durch schlechte Löslichkeit von Nintedanib bei neutralem pH-Wert verringert.
Nach Nahrungsaufnahme stieg die Nintedanib-Exposition gegenüber der Verabreichung im Nüchternzustand um etwa 20 % an (KI: 95,3-152,5 %) und die Resorption verlief verzögert (mediane tmax im Nüchternzustand: 2,00 h; nach Nahrungsaufnahme: 3,98 h).
Distribution
Nintedanib folgt einer zumindest biphasischen Verteilungskinetik. Nach intravenöser Infusion wurde ein hohes Verteilungsvolumen beobachtet (Vss: 1050 Liter).
Die Proteinbindung von Nintedanib in menschlichem Plasma war in vitro hoch (97,8 %). Es wird angenommen, dass die Bindung vorwiegend an Serumalbumin erfolgt. Nintedanib ist vorwiegend im Plasma verteilt mit einem Blut-Plasma-Verhältnis von 0,869.
Metabolismus
Hauptmetabolisierungsweg von Nintedanib ist eine hydrolytische Spaltung durch Esterasen, die zur Bildung der freien Säure BIBF 1202 führt. Anschliessend wird BIBF 1202 durch Enzyme (namentlich UGT 1A1, UGT 1A7, UGT 1A8 und UGT 1A10) in BIBF-1202-Glucuronid umgewandelt.
Nur ein kleiner Teil der Biotransformation von Nintedanib verläuft über CYP-Enzyme, vorwiegend über CYP 3A4. In der ADME-Studie entfiel der wesentliche Anteil der Radioaktivität im menschlichen Plasma auf Nintedanib (24 %), BIBF 1202 (32 %) und BIBF-1202-Glucuronid (30 %). In der ADME-Studie konnten die CYP-abhängigen Hauptmetaboliten im menschlichen Plasma nicht nachgewiesen werden. In vitro macht der CYP-abhängige Metabolismus etwa 5 % des Umsatzes aus, während unter den gegebenen experimentellen Bedingungen etwa 25 % auf die Esterspaltung entfielen.
Elimination
Die Gesamtplasmaclearance war nach intravenöser Infusion hoch (CL: 1390 ml/min). Innerhalb von 48 Stunden wurden nach oraler Gabe etwa 0,05 % und nach intravenöser Gabe etwa 1,4 % der Dosis als unveränderter Wirkstoff im Urin ausgeschieden. Die renale Clearance betrug 20 ml/min.
Ausscheidung
Nach oraler Verabreichung von [14C]-Nintedanib war der Haupteliminationsweg für arzneimittelgebundene Radioaktivität die Exkretion über Fäzes/Galle (93,4% der Dosis). Der grösste Teil von Ofev wurde in Form von BIBF 1202 über die Fäzes ausgeschieden (58% der Dosis); 20% wurden als Muttersubstanz und weniger als 7 % in Form von unterschiedlichen weniger wichtigen Metaboliten ausgeschieden. Der Beitrag der renalen Exkretion an der Gesamtclearance war niedrig (0,649% der Dosis). Die Wiederfindung wurde innerhalb von 4 Tagen nach der Verabreichung als vollständig bewertet (mehr als 90%). Die terminale Halbwertzeit von Nintedanib betrug 10-15 Stunden.
Beziehung Exposition/Ansprechen
An den Daten der Phase-II-Studie zur IPF durchgeführte explorative Analysen zur Beziehung zwischen Pharmakokinetik und unerwünschten Ereignissen zeigten, dass eine höhere Nintedanib-Exposition tendenziell mit einem Anstieg von Leberenzymen verbunden war (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Kinetik spezieller Patientengruppen, Intrinsische und extrinsische Faktoren
Den Ergebnissen einer populationspharmakokinetischen Analyse zu Patienten mit IPF und nicht-kleinzelligem Bronchialkarzinom (NSCLC) (N = 1191) sowie deskriptiven Untersuchungen zufolge hatten die Faktoren Geschlecht (für das Körpergewicht korrigiert), leichte und mittelschwere Einschränkung der Nierenfunktion (auf Grundlage der Creatinin-Clearance geschätzt), Alkoholkonsum und P-gp-Genotyp keinen Einfluss auf die Nintedanib-Exposition. Die populationspharmakokinetische Analyse zeigte einen moderaten Einfluss der weiter unten beschriebenen Faktoren Lebensalter, Körpergewicht und ethnische Abstammung auf die Nintedanib-Exposition. Vor dem Hintergrund der beobachteten hohen interindividuellen Variabilität kann aufgrund der moderaten Effekte dieser Kovariablen keine Empfehlung für eine Dosisanpassung gegeben werden (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Lebensalter
Die Nintedanib-Exposition stieg mit dem Alter linear an. Die AUCτ,ss fiel bei einem 45 Jahre alten Patienten (5. Perzentil) um 16 % niedriger und bei einem 76 Jahre alten Patienten (95. Perzentil) um 13 % höher aus als bei einem Patienten mit einem Alter von 62 Jahren (medianes Alter). Die Alterspanne der Analyse betrug 29 bis 85 Jahre und etwa 5 % der Population war älter als 75 Jahre.
Es wurden keine Studien zu Kindern und Jugendlichen durchgeführt.
Körpergewicht
Es wurde eine inverse Korrelation zwischen dem Körpergewicht und der Nintedanib-Exposition beobachtet. Die AUCτ,ss fiel bei einem 50 kg schweren Patienten (5. Perzentil) um 25 % höher und bei einem 100 kg schweren Patienten (95. Perzentil) um 19 % niedriger aus als bei einem Patienten mit einem Körpergewicht von 71,5 kg (medianes Körpergewicht).
Ethnische Abstammung
Der Populationsmittelwert der Nintedanib-Exposition war bei Chinesen, Taiwanern und Indern um 33-50 % höher und bei japanischen Patienten um 16 % höher, während er bei Koreanern um 16-22 % niedriger war als bei Patienten kaukasischer Abstammung (für das Körpergewicht korrigiert). Daten zu schwarzen Patienten liegen nur sehr begrenzt vor, bewegen sich jedoch im gleichen Bereich wie bei Patienten kaukasischer Abstammung.
Raucher
In der populationspharmakokinetischen Analyse fiel die Nintedanib-Exposition bei aktuellen Rauchern um 21 % niedriger aus als bei ehemaligen Rauchern und lebenslangen Nichtrauchern. Ein Effekt dieses Ausmasses rechtfertigt keine Dosisanpassung.
Eingeschränkte Leberfunktion
In einer speziellen Phase-I-Studie mit Einzeldosis und verglichen mit gesunden Teilnehmern war die Nintedanib-Exposition basierend auf der Cmax und der AUC bei Freiwilligen mit leichter Einschränkung der Leberfunktion (Child-Pugh-Klasse A; 90%-KI 1,3 – 3,7 für Cmax bzw. 1,2 – 3,8 für AUC) um das 2,2-Fache erhöht. Bei Freiwilligen mit moderater Einschränkung der Leberfunktion (Child-Pugh-Klasse B) war, verglichen mit gesunden Freiwilligen, die Exposition um das 7,6-Fache basierend auf der Cmax (90%-KI 4,4 – 13,2) bzw. um das 8,7-Fache (90%-KI 5,7 – 13,1) basierend auf der AUC erhöht. Patienten mit schwerer Einschränkung der Leberfunktion (Child-Pugh-Klasse C) wurden nicht untersucht.
Eingeschränkte Nierenfunktion
Die Pharmakokinetik von Nintedanib wurde nicht in einer speziellen PK Studie in Patienten mit eingeschränkter Nierenfunktion untersucht. Den Ergebnissen der populationspharmakokinetischen Analyse zufolge, hatte eine leichte (CrCl: 60 bis 90 ml/min) oder moderate (CrCl: 30 bis 60 ml/min) Einschränkung der Nierenfunktion keinen Einfluss auf die Nintedanib-Exposition. Es liegen nur begrenzt Daten zu Patienten mit schwerer Einschränkung der Nierenfunktion vor (CrCl unterhalb von 30 ml/min).
Präklinische Daten
Allgemeine Toxikologie
Untersuchungen zur Einzeldosis-Toxizität an Ratten und Mäusen weisen auf ein niedriges Potential für eine akute Toxizität von Nintedanib hin. In Untersuchungen zur Toxizität bei wiederholter Gabe an Ratten zeigten die meisten Nebenwirkungen (z.B. Verdickung der Epiphysenfuge, Läsionen an den Schneidezähnen) einen Zusammenhang mit dem Wirkmechanismus von Nintedanib (d.h. VEGFR-2-Hemmung). Derartige Veränderungen sind von anderen VEGFR-2-Inhibitoren bekannt und können als Klasseneffekte betrachtet werden.
In Toxizitätsstudien bei wiederholter Gabe an Nicht-Nagetier-Spezies wurden Diarrhö und Erbrechen, begleitet von verringerter Futteraufnahme und Gewichtsabnahme beobachtet.
Bei Ratten, Hunden und Langschwanzmakaken (Cynomolgus-Affen) gab es keine Hinweise auf einen Anstieg von Leberenzymen. Lediglich bei Rhesus-Affen wurden leichte Anstiege der Leberenzyme beobachtet, die nicht auf schwerwiegende Nebenwirkungen wie eine Diarrhö zurückzuführen waren.
Mutagenität/ Karzinogenität
Untersuchungen zur Genotoxizität zeigten kein mutagenes Potential von Nintedanib.
Die 2-Jahres-Studien zur Kanzerogenität an Mäusen und Ratten lieferten keine Evidenz für ein kanzerogenes Potential von Nintedanib.
Reproduktionstoxizität
Bei Ratten wurden bei einer Exposition, die etwa 3,6- bis 7,2-mal niedriger als die maximal empfohlene Humandosis (MRHD) von 150 mg zweimal täglich war, embryofetale Letalität und teratogene Wirkungen beobachtet. Bei einer Exposition, die etwa 12- bis 18-mal niedriger als die MRHD war, wurden geringfügige Wirkungen auf die Entwicklung des Achsenskeletts und die Entwicklung der grossen Arterien festgestellt.
Bei Kaninchen wurden bei einer Exposition in Höhe des etwa 3-fachen der MRHD embryofetale Letalität und teratogene Wirkungen beobachtet, während bei einer geringeren Exposition in Höhe der MRHD von 150 mg zweimal täglich bereits eine weniger eindeutige Beeinträchtigung der embryofetalen Entwicklung des Achsenskeletts und des Herzens festgestellt wurde.
Eine Studie an Ratten zur männlichen Fertilität und frühen embryonalen Entwicklung bis zur Implantation zeigte keinen Einfluss auf die männliche Reproduktionsrate oder Fertilität bei Expositionen in Höhe des etwa 3-fachen der MRHD.
Bei Ratten wurden geringe Mengen an radioaktiv markiertem Nintedanib und/oder seinen Metaboliten in die Milch ausgeschieden (≤0,5 % der verabreichten Dosis).
Sonstige Hinweise
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Ausser Reichweite von Kindern aufbewahren.
Bei Raumtemperatur (15-25°C) lagern. In der Originalverpackung aufbewahrt, um den Inhalt vor Feuchtigkeit zu schützen.
Zulassungsnummer
65330 (Swissmedic)
Zulassungsinhaberin
Boehringer Ingelheim (Schweiz) GmbH, Basel.
Stand der Information
Oktober 2020
Composizione
Principi attivi
Nintedanib (come nintedanib esilato)
Sostanze ausiliarie
- Contenuto della capsula: trigliceridi a catena media, grasso solido, lecitina (soia) (E322)
- Capsula: gelatina, glicerolo (85%), titanio diossido (E171), ferro ossido giallo (E172), ferro ossido rosso (E172)
- Inchiostro: gomma lacca, ferro ossido nero (E172), glicole propilenico (E1520)
Forma farmaceutica e quantità di principio attivo per unità
Capsule molli da 100 mg e 150 mg di nintedanib (equivalenti a 120,40 mg e 180,60 mg di nintedanib esilato)
Indicazioni/Possibilità d'impiego
Ofev è indicato per il trattamento
- della fibrosi polmonare idiopatica (IPF)
- di malattie interstiziali polmonari (ILD) fibrosanti croniche con fenotipo progressivo (vedere la rubrica «Efficacia clinica»)
- della malattia interstiziale polmonare associata a sclerosi sistemica (SSc-ILD)
Posologia/Impiego
Il trattamento deve essere iniziato da medici esperti nella diagnosi e nella terapia delle malattie per cui Ofev è indicato.
La dose raccomandata di Ofev è di 150 mg 2 volte al giorno ad intervalli di circa 12 ore.
Le capsule di Ofev devono essere assunte durante i pasti e deglutite intere con acqua. Non devono essere masticate né frantumate.
Se viene dimenticata una dose di Ofev, è necessario proseguire il trattamento con la successiva dose raccomandata all'orario previsto. Se viene saltata una dose, il paziente non deve prendere una dose aggiuntiva. La dose massima giornaliera raccomandata di 300 mg non deve essere superata.
Aggiustamento della dose
La gestione degli effetti indesiderati (vedere le rubriche «Avvertenze e misure precauzionali», «Effetti indesiderati») di Ofev include, oltre ad una terapia sintomatica (se indicata), una riduzione della dose o una sospensione temporanea del trattamento finché l'effetto collaterale non scompare o non ritorna a livelli che consentano il proseguimento della terapia. Il trattamento con Ofev può essere ripreso alla dose intera (150 mg due volte al giorno) o a una dose ridotta (100 mg due volte al giorno). Se il paziente non tollera la dose di 100 mg, il trattamento con Ofev deve essere interrotto.
In caso di sospensione del trattamento dovuta ad un aumento delle transaminasi (AST o ALT) superiore a 3 volte il limite superiore del valore normale (ULN), dopo che i valori delle transaminasi si sono normalizzati è possibile riprendere il trattamento con Ofev a una dose ridotta (100 mg due volte al giorno) e, successivamente, aumentarla alla dose intera (150 mg due volte al giorno). In caso di aumento di AST o ALT superiore a 5 volte l'ULN oppure di aumento superiore a 3 volte l'ULN e di contemporanei riscontri o sintomi di una compromissione epatica severa, è necessario interrompere Ofev (vedere le rubriche «Avvertenze e misure precauzionali», «Effetti indesiderati»).
Istruzioni posologiche speciali
Pazienti con disturbi della funzionalità renale
Meno dell'1% di una dose singola di nintedanib è escreto attraverso i reni (vedere la rubrica «Farmacocinetica»). Non è necessario l'aggiustamento della dose iniziale nei pazienti con compromissione renale da lieve a moderata. La sicurezza, l'efficacia e la farmacocinetica di nintedanib non sono state studiate nei pazienti con compromissione renale severa (CrCL <30 ml/min).
Pazienti con disturbi della funzionalità epatica
Nintedanib viene eliminato prevalentemente per escrezione biliare/fecale (> 90%; vedere la rubrica «Farmacocinetica»). L'esposizione è aumentata nei pazienti con compromissione epatica (classe Child-Pugh A, classe Child-Pugh B). Nei pazienti con compromissione epatica lieve (classe Child-Pugh A) la dose raccomandata di Ofev è di 100 mg due volte al giorno, a circa 12 ore di distanza. Nei pazienti con compromissione epatica lieve (classe Child-Pugh A) deve essere presa in considerazione la sospensione temporanea o l'interruzione del trattamento per la gestione delle reazioni avverse.
La sicurezza e l'efficacia di nintedanib non sono state studiate nei pazienti con compromissione epatica di classe Child Pugh B o C. L'uso di Ofev nei pazienti con compromissione epatica moderata (classe Child Pugh B) o severa (classe Child Pugh C) non è raccomandato (vedere le rubriche «Avvertenze e misure precauzionali» e «Farmacocinetica»).
Bambini e adolescenti
La sicurezza e l'efficacia di Ofev nei bambini e negli adolescenti non sono state esaminate in studi clinici.
Pazienti anziani (≥65 anni)
Nei pazienti anziani non sono state osservate differenze nella sicurezza ed efficacia generale rispetto ai pazienti di età inferiore a 65 anni. Non è necessario alcun aggiustamento della dose in funzione dell'età del paziente (vedere la rubrica «Farmacocinetica»).
Controindicazioni
Ofev è controindicato nei pazienti con ipersensibilità nota a nintedanib, alle arachidi, alla soia o ad uno qualsiasi degli altri componenti.
Ofev è controindicato in gravidanza (vedere le rubriche «Gravidanza/allattamento» e «Dati preclinici»).
Avvertenze e misure precauzionali
Patologie gastrointestinali
Diarrea
Negli studi clinici (vedere la rubrica «Efficacia clinica») la diarrea è stata la reazione avversa gastrointestinale più frequente.
Nella maggior parte dei pazienti la diarrea è stata di intensità da lieve a moderata e si è manifestata entro i primi 3 mesi di trattamento.
Negli studi INPULSIS (pazienti con IPF) la diarrea è stata riportata nel 62,4% dei pazienti trattati con Ofev rispetto al 18,4% dei pazienti trattati con placebo. La diarrea ha causato la riduzione della dose di Ofev nel 10,7% dei pazienti rispetto a nessuno dei pazienti trattati con placebo e l'interruzione del trattamento con Ofev nel 4,4% dei pazienti rispetto allo 0,2% dei pazienti trattati con placebo.
Nello studio INBUILD (pazienti con ILD fibrosanti progressive) la diarrea è stata riportata nel 66,9% dei pazienti trattati con Ofev rispetto al 23,9% dei pazienti trattati con placebo. La diarrea ha determinato la riduzione della dose di Ofev nel 16,0% dei pazienti rispetto allo 0,9% dei pazienti trattati con placebo e l'interruzione del trattamento con Ofev nel 5,7% dei pazienti rispetto allo 0,3% dei pazienti trattati con placebo.
Nello studio SENSCIS (pazienti con SSc-ILD) la diarrea è stata riportata nel 75,7% dei pazienti trattati con Ofev rispetto al 31,6% dei pazienti trattati con placebo.
La diarrea ha determinato la riduzione della dose di Ofev nel 22,2% dei pazienti rispetto all'1,0% dei pazienti trattati con placebo e l'interruzione del trattamento nel 6,9% dei pazienti rispetto allo 0,3% dei pazienti trattati con placebo (vedere la rubrica «Effetti indesiderati»).
La diarrea può portare a disidratazione associata o meno a disturbi elettrolitici, con conseguenti possibili disturbi della funzionalità renale.
Alla comparsa dei primi segni di diarrea, i pazienti devono essere trattati con un'adeguata terapia reidratante e con la somministrazione di antidiarroici, ad esempio loperamide. Ciò può richiedere, se necessario, la riduzione della dose o l'interruzione del trattamento. Il trattamento con Ofev può essere ripreso a una dose ridotta (100 mg due volte al giorno) o alla dose intera (150 mg due volte al giorno). In caso di diarrea severa persistente nonostante la terapia sintomatica, il trattamento con Ofev deve essere interrotto.
Nausea e vomito
Nausea e vomito sono eventi avversi segnalati di frequente (vedere la rubrica «Effetti indesiderati») e per lo più con intensità da lieve a moderata.
Negli studi INPULSIS (pazienti con IPF) la nausea è stata riportata nel 24,5% dei pazienti trattati con Ofev rispetto al 6,6% dei pazienti trattati con placebo. La nausea ha determinato la riduzione della dose di Ofev nell'1,7% dei pazienti rispetto a nessuno dei pazienti trattati con placebo e ha causato l'interruzione del trattamento con Ofev nel 2,0% dei pazienti rispetto a nessuno dei pazienti trattati con placebo.
Nello studio INBUILD (pazienti con ILD fibrosanti progressive) la nausea è stata riportata nel 28,9% dei pazienti trattati con Ofev rispetto al 9,4% dei pazienti trattati con placebo. La nausea ha determinato la riduzione della dose di Ofev nel 3,3% dei pazienti rispetto allo 0,6% dei pazienti trattati con placebo e l'interruzione del trattamento con Ofev nello 0,3% dei pazienti rispetto allo 0,3% dei pazienti trattati con placebo.
Nello studio SENSCIS (pazienti con SSc-ILD) la nausea è stata riportata nel 31,6% dei pazienti trattati con Ofev rispetto al 13,5% dei pazienti trattati con placebo. La nausea ha determinato la riduzione della dose di Ofev nel 2,1% dei pazienti rispetto a nessuno dei pazienti trattati con placebo e l'interruzione del trattamento con Ofev nel 2,1% dei pazienti rispetto a nessuno dei pazienti trattati con placebo.
Negli studi INPULSIS (pazienti con IPF) il vomito è stato riportato nell'11,6% dei pazienti trattati con Ofev rispetto al 2,6% dei pazienti trattati con placebo. Il vomito ha determinato la riduzione della dose di Ofev nell'1,1% dei pazienti rispetto a nessuno dei pazienti trattati con placebo e l'interruzione del trattamento con Ofev nello 0,8% dei pazienti rispetto a nessuno dei pazienti trattati con placebo.
Nello studio INBUILD (pazienti con ILD fibrosanti progressive) il vomito è stato riportato nel 18,4% dei pazienti trattati con Ofev rispetto al 5,1% dei pazienti trattati con placebo. Il vomito ha determinato la riduzione della dose di Ofev nel 2,4% dei pazienti rispetto allo 0,9% dei pazienti trattati con placebo e l'interruzione del trattamento con Ofev nello 0,9% dei pazienti rispetto a nessuno dei pazienti trattati con placebo.
Nello studio SENSCIS (pazienti con SSc-ILD) il vomito è stato riportato nel 24,7% dei pazienti trattati con Ofev rispetto al 10,4% dei pazienti trattati con placebo. Il vomito ha determinato la riduzione della dose di Ofev nel 2,1% dei pazienti rispetto a nessuno dei pazienti trattati con placebo e l'interruzione del trattamento con Ofev nell'1,4% dei pazienti rispetto allo 0,3% dei pazienti trattati con placebo.
Il vomito può portare a disidratazione associata o meno a disturbi elettrolitici, con conseguenti possibili disturbi della funzionalità renale.
Se i sintomi persistono nonostante una terapia di supporto appropriata (inclusa la terapia antiemetica), può essere necessario ridurre la dose o interrompere il trattamento. Il trattamento può essere ripreso a una dose ridotta (100 mg due volte al giorno) o alla dose intera (150 mg due volte al giorno). In caso di sintomi severi e persistenti il trattamento con Ofev deve essere interrotto.
Funzionalità epatica
La sicurezza e l'efficacia di Ofev non sono state studiate nei pazienti con compromissione epatica moderata (classe Child-Pugh B) o severa (classe Child-Pugh C). Pertanto l'uso di Ofev non è raccomandato in questi pazienti.
A causa dell'esposizione aumentata, il rischio di eventi avversi può essere aumentato nei pazienti con compromissione epatica lieve (classe Child-Pugh A). I pazienti con compromissione epatica lieve (classe Child Pugh A) devono essere trattati con una dose ridotta di Ofev (vedere le rubriche «Posologia/impiego» e «Farmacocinetica»).
Nei pazienti trattati con nintedanib sono stati osservati casi di danno epatico indotto da medicamenti. Durante l'osservazione post-marketing sono stati osservati sia casi non gravi che casi gravi di danno epatico indotto da medicamenti, anche con esito fatale.
Nella maggior parte dei casi, gli eventi avversi epatici si sono verificati entro i primi tre mesi di trattamento. Pertanto, i livelli epatici di transaminasi e bilirubina devono essere misurati prima dell'inizio del trattamento con Ofev, ad intervalli regolari durante i primi tre mesi di trattamento e successivamente a cadenza periodica (ad es. ad ogni visita del paziente) oppure secondo indicazione clinica.
Nella maggior parte dei casi, gli aumenti dei valori degli enzimi epatici (ALT, AST, ALKP, gamma-glutamil transferasi (GGT)) e della bilirubina erano reversibili a seguito di riduzione della dose o interruzione del trattamento. Valori ALT e/o AST elevati, che sono aumentati di almeno 3 volte il limite superiore del valore normale (ULN), sono stati osservati fino al 5,0%, al 7,8% o al 4,9% dei pazienti trattati con Ofev negli studi INPULSIS (pazienti con IPF), INBUILD (pazienti con ILD fibrosanti progressive) o SENSCIS (pazienti con SSc-ILD), mentre complessivamente in meno del 2% dei pazienti trattati con placebo.
In caso di aumento delle transaminasi (AST o ALT) superiore a 3 volte il limite superiore del valore normale (ULN), si raccomanda di ridurre la dose a 100 mg due volte al giorno o di interrompere il trattamento con Ofev. Inoltre, il paziente deve essere monitorato scrupolosamente e devono essere ricercate eventuali cause alternative dell'aumento degli enzimi epatici. Dopo la normalizzazione dei valori delle transaminasi, il trattamento con Ofev può essere ripreso a una dose ridotta (100 mg due volte al giorno) che, successivamente, può essere aumentata alla dose intera (150 mg due volte al giorno). Se l'aumento dei valori epatici è associato a segni clinici di danno epatico, ad es. ittero, oppure se le transaminasi (AST o ALT) aumentano più di 5 volte l'ULN, il trattamento con Ofev deve essere interrotto in modo permanente (vedere la rubrica «Posologia/impiego»).
I pazienti con basso peso corporeo (< 65 kg), di etnia asiatica e di sesso femminile sono esposti a un rischio più elevato di aumento degli enzimi epatici.
L'esposizione a nintedanib aumenta in modo lineare con l'età del paziente. Ciò potrebbe causare anche un maggiore rischio di aumento degli enzimi epatici (vedere la rubrica «Farmacocinetica»).
Si raccomanda un monitoraggio scrupoloso dei pazienti che presentano questi fattori di rischio.
Funzionalità renale
Con l'uso di nintedanib sono stati segnalati casi di compromissione o insufficienza renale, alcuni dei quali con esito fatale (vedere la rubrica «Effetti indesiderati»).
Durante la terapia con nintedanib si raccomanda di monitorare i pazienti, in particolare quelli che presentano fattori di rischio di compromissione o insufficienza renale. In caso di compromissione o insufficienza renale deve essere preso in considerazione un aggiustamento della terapia (vedere la rubrica «Aggiustamento della dose»).
Sanguinamenti
Dato il suo meccanismo d'azione (inibizione del recettore del fattore di crescita dell'endotelio vascolare (vascular endothelial growth factor receptor, VEGFR), Ofev può essere associato ad aumento del rischio di sanguinamento.
Negli studi clinici con Ofev, la frequenza degli eventi emorragici è risultata leggermente superiore nei pazienti trattati con Ofev e/o simile tra i gruppi di trattamento (Ofev 10,3% rispetto a placebo 7,8% negli studi INPULSIS (pazienti con IPF); Ofev 11,1% rispetto a placebo 12,7% nello studio INBUILD (pazienti con ILD fibrosanti progressive); Ofev 11,1% rispetto a placebo 8,3% nello studio SENSCIS (pazienti con SSc-ILD)).
L'evento emorragico più frequente è stato epistassi non grave.
La frequenza degli eventi emorragici gravi è stata ridotta in entrambi i gruppi di trattamento (Ofev 1,3% rispetto a placebo 1,4% nello studio INPULSIS (pazienti con IPF); Ofev 0,9% rispetto a placebo 1,5% nello studio INBUILD (pazienti con ILD fibrosanti progressive); Ofev 1,4% rispetto a placebo 0,7% nello studio SENSCIS (pazienti con SSc-ILD)).
Non sono stati inclusi negli studi clinici pazienti con rischio noto di sanguinamento, ad esempio pazienti con predisposizione ereditaria al sanguinamento, né pazienti in trattamento con una dose intera di anticoagulante. Nel periodo post-marketing sono stati riportati eventi emorragici, alcuni dei quali gravi e fatali (inclusi pazienti sottoposti o meno a terapia anticoagulante oppure trattati con altri medicamenti che possono provocare sanguinamento). Pertanto, i pazienti con fattori di rischio supplementari devono essere trattati con Ofev solo se i benefici attesi superano il rischio potenziale.
Eventi tromboembolici arteriosi
I pazienti con storia recente di infarto miocardico o ictus sono stati esclusi dagli studi clinici.
Negli studi clinici gli eventi tromboembolici arteriosi sono stati segnalati con una frequenza ridotta (Ofev 2,5% rispetto a placebo 0,7% negli studi INPULSIS (pazienti con IPF); Ofev 0,9% rispetto a placebo 0,9% nello studio INBUILD (pazienti con ILD fibrosanti progressive); Ofev 0,7% rispetto a placebo 0,7% nello studio SENSCIS (pazienti con SSc-ILD)). Negli studi INPULSIS sono stati segnalati casi di infarto miocardico con una frequenza maggiore nei pazienti del gruppo Ofev (1,6%) rispetto ai pazienti del gruppo placebo (0,5%), mentre eventi avversi riconducibili a patologia cardiaca ischemica si sono verificati con frequenza simile nei gruppi Ofev e placebo.
Nello studio INBUILD e SENSCIS è stata osservata una bassa frequenza di infarto miocardico: Ofev 0,9% rispetto a placebo 0,9% nello studio INBUILD; Ofev 0% rispetto a placebo 0,7% nello studio SENSCIS.
È richiesta cautela nel trattamento di pazienti con aumentato rischio cardiovascolare, ad esempio in quelli con patologie coronariche note. Nei pazienti che sviluppano segni o sintomi di ischemia miocardica acuta deve essere presa in considerazione l'interruzione del trattamento.
Aneurismi e dissezioni arteriose
L'uso di inibitori del pathway del VEGF in pazienti con o senza ipertensione può favorire la formazione di aneurismi e/o dissezioni arteriose. Prima di iniziare il trattamento con Ofev, questo rischio deve essere attentamente considerato in pazienti con fattori di rischio quali ipertensione o storia anamnestica di aneurisma.
Tromboembolismo venoso
Negli studi clinici non è stato osservato un aumento del rischio di tromboembolismo venoso nei pazienti trattati con nintedanib. Dato il meccanismo d'azione di nintedanib, i pazienti possono essere esposti a un maggiore rischio di eventi tromboembolici.
Perforazioni gastrointestinali
Dato il meccanismo d'azione di nintedanib, i pazienti trattati con questo medicamento possono presentare un maggiore rischio di perforazione gastrointestinale. Negli studi INPULSIS (pazienti con IPF) i sono state riportate perforazioni gastrointestinali nello 0,3% dei pazienti trattati con Ofev e in nessuno (0) dei pazienti trattati con placebo. Nello studio SENSCIS (pazienti con SSc-ILD) e nello studio INBUILD (pazienti con ILD fibrosanti progressive) non sono stati segnali casi di perforazione gastrointestinale in nessuno dei gruppi di trattamento.
Nel periodo post-marketing sono stati riportati casi di perforazioni gastrointestinali, alcuni dei quali fatali. È richiesta particolare cautela nel trattamento di pazienti sottoposti a recenti interventi di chirurgia addominale, con recente perforazione di un organo cavo, precedenti anamnestici di ulcera peptica o malattia diverticolare oppure in trattamento concomitante con corticosteroidi o FANS.
Il trattamento con Ofev deve essere interrotto in modo permanente nei pazienti che sviluppano perforazione gastrointestinale.
I pazienti con rischio noto di perforazione gastrointestinale possono essere trattati con Ofev solo se i benefici attesi superano il rischio potenziale.
Compromissione della guarigione delle ferite
Negli studi clinici non è stato osservato un aumento dell'incidenza della compromissione della guarigione delle ferite. Dato il suo meccanismo d'azione, nintedanib può compromettere la guarigione delle ferite. Non sono stati condotti studi specifici per indagare l'effetto di nintedanib sulla guarigione delle ferite. Pertanto, il trattamento con Ofev deve essere iniziato, oppure ripreso in caso di interruzione perioperatoria, in base al giudizio clinico riguardo ad un'adeguata guarigione della ferita.
Lecitina di soia
Le capsule molli di Ofev contengono lecitina di soia (vedere la rubrica «Controindicazioni»).
Interazioni
Sono stati condotti studi su potenziali interazioni esclusivamente in soggetti adulti.
Influenza di altri principi attivi su nintedanib:
Inibitori e induttori della glicoproteina P (P-gp) e del CYP3A4
Nintedanib è un substrato della P-gp (vedere la rubrica «Farmacocinetica») e, in misura ridotta, anche del CYP3A4. La co-somministrazione con ketoconazolo, un potente inibitore della P-gp e un inibitore del CYP3A4, ha aumentato l'AUC di nintedanib di 1,61 volte e la Cmax di 1,83 volte.
Se co-somministrati con Ofev, alcuni potenti inibitori della P-gp (ad es. ketoconazolo o eritromicina) possono aumentare l'esposizione a nintedanib. In questi casi i pazienti devono essere monitorati scrupolosamente per verificare la tollerabilità di nintedanib. La gestione degli effetti indesiderati può richiedere la sospensione, la riduzione della dose o l'interruzione del trattamento con Ofev (vedere la rubrica «Posologia/impiego»).
La co-somministrazione con rifampicina, un potente induttore della P-gp e del CYP3A4, ha aumentato l'AUC di nintedanib al 50,3% e la Cmax al 60,3% rispetto al valore misurato per nintedanib in monoterapia.
I potenti induttori della P-gp (ad es. rifampicina, carbamazepina, fenitoina ed erba di San Giovanni) possono diminuire l'esposizione a nintedanib. Deve essere valutata la scelta di un medicamento alternativo da co-somministrare con nintedanib che abbia un potenziale di induzione della P-gp minimo o assente.
Assunzione con cibo
Ofev deve essere assunto con cibo per aumentarne la biodisponibilità (vedere la rubrica «Farmacocinetica»).
pH
La solubilità di nintedanib dipende notevolmente dal pH e diminuisce in presenza di un pH >3. Negli studi clinici, la co-somministrazione di medicamenti che aumentano il pH gastrico non ha influenzato i valori Ctrough di nintedanib in caso di assunzione di nintedanib con cibo. In caso di assunzione a digiuno, la co-somministrazione di medicamenti che aumentano il pH gastrico potrebbe ridurre la biodisponibilità di nintedanib. Si raccomanda pertanto di assumere nintedanib con cibo.
Fumo
Il fumo è correlato ad una ridotta esposizione a nintedanib, quindi potrebbe alterarne l'efficacia. I pazienti dovrebbero essere incoraggiati a smettere di fumare prima di iniziare la terapia con Ofev o a ridurre il fumo durante la terapia.
Secondo test in vitro, nintedanib non è un substrato di OATP-1B1, OATP-1B3, OATP-2B1, OCT-2, BCRP o MRP-2. Studi in vitro hanno mostrato che nintedanib è un substrato di OCT-1. Si suppone, tuttavia, che queste osservazioni abbiano rilevanza clinica ridotta.
Influenza di nintedanib su altri principi attivi:
Trasportatori
In vitro è stato osservato un debole potenziale inibitorio su OCT-1, BCRP e P-gp. La relativa rilevanza clinica non è stata studiata, ma viene comunque ritenuta ridotta.
Secondo test in vitro, nintedanib non è un inibitore di OATP-1B1, OATP-1B3, OATP-2B1, OCT-2 o MRP-2.
Il metabolita BIBF 1202 è risultato in vitro un debole inibitore di OATP-1B1, OATP-1B3, OATP-2B1, OCT-1 e BCRP. La relativa rilevanza clinica non è stata studiata, ma viene comunque ritenuta ridotta.
Enzimi del citocromo (CYP)
Solo una minima parte della biotrasformazione di nintedanib dipende dagli enzimi del CYP. Negli studi preclinici, nintedanib e i suoi metaboliti, BIBF 1202 con il gruppo funzionale acido libero e il suo glucuronide, non hanno inibito né indotto gli enzimi del CYP (vedere la rubrica «Farmacocinetica»). Si ritiene che la probabilità di interazioni medicamentose con nintedanib sulla base del metabolismo del CYP sia bassa.
Co-somministrazione con altri medicamenti
Terapia concomitante con pirfenidone:
Sulla base dei risultati di uno speciale studio di farmacocinetica (FC), non vi è alcuna evidenza di un'interazione farmacocinetica rilevante tra nintedanib e pirfenidone somministrati in associazione.
Terapia concomitante con bosentan:
In uno studio di farmacocinetica (FC) dedicato è stato valutato il trattamento con Ofev in concomitanza con bosentan su volontari sani. I partecipanti allo studio hanno ricevuto una dose singola di 150 mg di Ofev prima e dopo dosi multiple da 125 mg di bosentan due volte al giorno allo stato stazionario. Le percentuali geometriche medie aggiustate (intervallo di confidenza [IC] 90%) erano pari al 103% (86% - 124%) o al 99% (91% - 107%) rispettivamente per la Cmax e l'AUC0-tz di nintedanib (n=13), a conferma che la somministrazione di nintedanib in concomitanza con bosentan non ha alterato la farmacocinetica di nintedanib.
Le potenziali interazioni tra nintedanib e i contraccettivi ormonali non sono state studiate.
Gravidanza/Allattamento
Gravidanza
Non sono disponibili dati clinici sull'utilizzo di Ofev in gravidanza, tuttavia studi preclinici sugli animali hanno mostrato una tossicità per la riproduzione di questo medicamento (vedere la rubrica «Dati preclinici»). Nintedanib può causare danni al feto nell'uomo, quindi non deve essere usato durante la gravidanza.
Prima di iniziare il trattamento con Ofev deve essere effettuato un test di gravidanza, da ripetersi durante il trattamento in caso di necessità.
Le pazienti devono essere invitate ad informare il loro medico o il loro farmacista nel caso in cui avviassero una gravidanza durante il trattamento con Ofev.
Se una paziente avvia una gravidanza durante il trattamento con Ofev, il trattamento deve essere interrotto e la paziente deve essere informata sui potenziali rischi per il feto.
Contraccezione
Nintedanib può causare danni al feto (vedere la rubrica «Dati preclinici»). Le donne in età potenzialmente fertile che vengono trattate con Ofev devono essere avvisate di non avviare una gravidanza durante il trattamento con Ofev e di usare metodi contraccettivi altamente efficaci durante tutto il trattamento e per almeno 3 mesi dall'ultima dose di Ofev.
Al momento non è noto se nintedanib possa ridurre o meno l'efficacia dei contraccettivi ormonali e pertanto le donne che fanno uso di contraccettivi ormonali devono utilizzare anche un metodo barriera.
Allattamento
Non esistono informazioni sull'escrezione di nintedanib e dei suoi metaboliti nel latte materno.
Gli studi preclinici hanno mostrato che piccole quantità di nintedanib e dei suoi metaboliti (≤0,5% della dose somministrata) sono escrete nel latte delle ratte in allattamento.
Un rischio per i neonati/lattanti non può essere escluso. L'allattamento deve essere interrotto durante il trattamento con Ofev.
Effetti sulla capacità di condurre veicoli e sull'impiego di macchine
Non sono stati effettuati studi sulla capacità di guidare veicoli o sulla capacità di utilizzare macchine.
I pazienti devono essere invitati a osservare cautela nella guida di veicoli o nell'uso di macchine durante il trattamento con Ofev.
Effetti indesiderati
La sicurezza di Ofev è stata esaminata in studi clinici su più di 1.000 pazienti con IPF, di cui più di 200 sono stati trattati con Ofev per una durata superiore a 2 anni.
Ofev è stato esaminato su pazienti con IPF in tre studi clinici randomizzati, controllati con placebo, in doppio cieco della durata di 52 settimane. Nello studio di fase II (TOMORROW) e negli studi di fase III (INPULSIS-1 e INPULSIS-2), 723 pazienti con IPF sono stati trattati con Ofev 150 mg due volte al giorno e 508 pazienti con placebo. La durata mediana dell'esposizione è stata di 10 mesi per i pazienti trattati con Ofev, mentre di 11 mesi per i pazienti trattati con placebo. I partecipanti agli studi avevano un'età compresa tra 42 e 89 anni (età mediana di 67 anni). I pazienti erano in gran parte uomini (79%) e di origine caucasica (60%).
Ofev è stato inoltre esaminato in uno studio di fase III, randomizzato, in doppio cieco, controllato con placebo (INBUILD), che ha confrontato il trattamento con Ofev 150 mg due volte al giorno (n=332) con placebo (n=331) in 663 pazienti con altre ILD fibrosanti croniche con fenotipo progressivo. I pazienti sono stati trattati per almeno 52 settimane, ma alcuni pazienti hanno proseguito il trattamento fino a 27 mesi. La durata mediana dell'esposizione è stata di 16 mesi per i pazienti trattati con Ofev, mentre di 17 mesi per i pazienti trattati con placebo. I partecipanti allo studio avevano un'età compresa tra 27 e 87 anni (età mediana: 67 anni).). I pazienti erano in gran parte uomini (54%), di origine caucasica (74%) o asiatica (25%).
Ofev è stato esaminato in uno studio di fase III (SENSCIS), randomizzato, in doppio cieco, controllato verso placebo, in cui 576 pazienti con SSc-ILD sono stati trattati con 150 mg di Ofev due volte al giorno (n=288) o placebo (n=288). I pazienti sono stati trattati per almeno 52 settimane, ma alcuni pazienti hanno proseguito il trattamento fino a 100 settimane. La durata di esposizione mediana è stata di 15 mesi nei pazienti trattati con Ofev e di 16 mesi nei pazienti trattati con placebo. L'età dei volontari era compresa tra 20 e 79 anni (età mediana 55 anni). I pazienti erano in gran parte di sesso femminile (75%). I pazienti erano prevalentemente di etnia caucasica (67%), asiatici (25%) o nera (6%). Il 49% dei pazienti era stabilmente in terapia con micofenolato al basale.
Gli effetti indesiderati segnalati con maggiore frequenza in associazione all'uso di Ofev includevano diarrea, nausea e vomito, dolore addominale, diminuzione dell'appetito, diminuzione del peso, sanguinamenti, aumento degli enzimi epatici ed eruzione cutanea.
Il profilo di sicurezza nei pazienti trattati con Ofev è risultato simili per i pazienti con o senza micofenolato al basale.
La rubrica «Avvertenze e misure precauzionali» contiene indicazioni sulla procedura da adottare in relazione ai rispettivi effetti indesiderati.
Per quanto riguarda la frequenza degli effetti indesiderati, sono state adottate le categorie di frequenza secondo la convenzione seguente: «molto comune» (≥1/10), «comune» (< 1/10, ≥1/100), «non comune» (< 1/100, ≥1/1000), «raro» (< 1/1000, ≥1/10'000), «molto raro» (< 1/10'000).
Fibrosi polmonare idiopatica (IPF)
Patologie del sistema emolinfopoietico
Non comune: Trombocitopenia.
Disturbi del metabolismo e della nutrizione
Comune: Diminuzione dell'appetito, diminuzione del peso.
Patologie del sistema nervoso
Comune: Cefalea.
Patologie vascolari
Comune: Sanguinamentia.
Non comune: Ipertensioneb.
Frequenza non nota: Aneurismi e dissezioni arteriose.
Patologie gastrointestinali
Molto comune: Diarrea (53,6%), nausea (19,1%), dolore addominalec (10,2%).
Comune: Vomito.
Non comune: Pancreatite, perforazione gastrointestinale, colite.
Patologie epatobiliari
Molto comune: Aumento degli enzimi epatici (10,5%).
Comune: Aumento dell'alanina aminotransferasi (ALT), aumento dell'aspartato aminotransferasi (AST), aumento della gamma-glutamil transferasi (GGT) .
Non comune: Aumento della fosfatasi alcalina ematica (ALP), iperbilirubinemia, danno epatico indotto da medicamenti.
Patologie della cute e del tessuto sottocutaneo
Comune: Eruzione cutanea.
Non comune: Prurito, alopecia.
Patologie renali e urinarie
Frequenza non nota: Insufficienza renale (vedere la rubrica «Avvertenze e misure precauzionali»).
a Nel periodo post-marketing sono stati osservati eventi emorragici non gravi e gravi, alcuni anche con esito fatale, che sono coerenti con i dati esperienziali degli studi clinici. Gli eventi emorragici più frequenti hanno incluso sanguinamento rettale/ematochezia, epistassi ed ematomi. Gli eventi fatali hanno riguardato sanguinamenti gastrointestinali, intracranici e polmonari, nonché DIC.
b Include ipertensione, pressione arteriosa elevata, crisi ipertensiva e cardiomiopatia ipertensiva.
c Include dolore addominale, dolore all'addome superiore, dolore all'addome inferiore, dolori gastrointestinali, dolore addominale da compressione.
Altre malattie interstiziali polmonari (ILD) fibrosanti croniche con fenotipo progressivo
Patologie del sistema emolinfopoietico
Non comune: Trombocitopenia.
Disturbi del metabolismo e della nutrizione
Molto comune: Diminuzione dell'appetito (11,1%).
Comune: Diminuzione del peso.
Patologie del sistema nervoso
Comune: Cefalea.
Patologie vascolari
Comune: Ipertensione, sanguinamenti.
Frequenza non nota: Aneurismi e dissezioni arteriose.
Patologie gastrointestinali
Molto comune: Diarrea (59%), nausea (24%), dolore addominale (11%), vomito (12%).
Non comune: Pancreatite, colite.
Frequenza non nota: Perforazione gastrointestinale.
Patologie epatobiliari
Molto comune: Aumento degli enzimi epatici (18,4%), aumento dell'alanina aminotransferasi (ALT) (10,8%).
Comune: Aumento dell'aspartato aminotransferasi (AST), aumento della gamma-glutamil transferasi (GGT), aumento della fosfatasi alcalina ematica (ALP), danno epatico indotto da medicamenti.
Non comune: Iperbilirubinemia.
Patologie della cute e del tessuto sottocutaneo
Comune: Eruzione cutanea.
Non comune: Prurito, alopecia.
Patologie renali e urinarie
Frequenza non nota: Insufficienza renale (vedere la rubrica «Avvertenze e misure precauzionali»).
Malattia interstiziale polmonare associata a sclerosi sistemica (SSc-ILD)
Patologie del sistema emolinfopoietico
Non comune: Trombocitopenia.
Disturbi del metabolismo e della nutrizione
Comune: Diminuzione dell'appetito, diminuzione del peso.
Patologie del sistema nervoso
Comune: Cefalea.
Patologie vascolari
Comune: Ipertensione, sanguinamenti.
Frequenza non nota: Aneurismi e dissezioni arteriose.
Patologie gastrointestinali
Molto comune: Diarrea (68,4%), nausea (24,7%), dolore addominale (11,8%), vomito (17,7%).
Non comune: Colite.
Frequenza non nota: Pancreatite, perforazione gastrointestinale.
Patologie epatobiliari
Molto comune: Aumento degli enzimi epatici (11,1%).
Comune: Aumento dell'alanina aminotransferasi (ALT), aumento dell'aspartato aminotransferasi (AST), aumento della gamma-glutamil transferasi (GGT), aumento della fosfatasi alcalina ematica (ALP) .
Non comune: Danno epatico indotto da medicamenti.
Frequenza non nota: Iperbilirubinemia.
Patologie della cute e del tessuto sottocutaneo
Non comune: Eruzione cutanea, prurito.
Frequenza non nota: Alopecia.
Patologie renali e urinarie
Non comune: Insufficienza renale (vedere la rubrica «Avvertenze e misure precauzionali»).
La notifica di effetti collaterali sospetti dopo l'omologazione del medicamento è molto importante. Consente una sorveglianza continua del rapporto rischio-benefico del medicamento. Chi esercita una professione sanitaria è invitato a segnalare qualsiasi nuovo o grave effetto collaterale sospetto attraverso il portale online ElViS (Electronic Vigilance System). Maggiori informazioni sul sito www.swissmedic.ch.
Posologia eccessiva
Non esiste un antidoto specifico né un trattamento per il sovradosaggio da Ofev. La dose singola massima di nintedanib somministrata negli studi di fase I è stata di 450 mg una volta al giorno. Inoltre, 2 pazienti hanno ricevuto un sovradosaggio massimo di 600 mg due volte al giorno fino a otto giorni. Gli eventi avversi osservati erano coerenti con il profilo di sicurezza noto per nintedanib, cioè aumento degli enzimi epatici e sintomi gastrointestinali. Tali eventi si sono risolti in entrambi i pazienti.
Negli studi INPULSIS (pazienti con IPF), un paziente è stato inavvertitamente esposto ad una dose di 600 mg al giorno per un totale di 21 giorni. Durante il periodo di posologia errata si è verificato un evento avverso non grave (nasofaringite) risoltosi spontaneamente, mentre non sono insorti altri eventi.
In caso di sovradosaggio, il trattamento deve essere interrotto e devono essere avviate adeguate misure generali di supporto.
Proprietà/Effetti
Codice ATC
L01XE31
Meccanismo d'azione
Nintedanib è una piccola molecola che inibisce l'attività dei recettori tirosin-chinasici, tra cui il fattore di crescita derivato dalle piastrine (PDGFR) α e ß, il fattore di crescita dei fibroblasti (FGFR) 1-3 e il fattore di crescita dell'endotelio vascolare (VEGFR) 1-3. Nintedanib si lega in modo competitivo al sito di legame per l'adenosina trifosfato (ATP) di questi recettori e blocca la segnalazione intracellulare, che svolge un ruolo determinate nella proliferazione, migrazione e trasformazione dei fibroblasti, quali patomeccanismi essenziali dell'IPF. Inoltre, nintedanib inibisce le chinasi Flt-3 (proteina tirosin chinasi Fms-simile), Lck (proteina tirosin chinasi linfocito-specifica), Lyn (proteina tirosin chinasi lyn) e Src (proteina tirosin chinasi proto-oncogena src).
Farmacodinamica
Nintedanib inibisce l'attivazione delle cascate di segnali FGFR e PDGFR che sono determinanti nella proliferazione, migrazione e differenziazione dei fibroblasti/miofibroblasti polmonari, ossia le cellule caratteristiche della patologia della fibrosi polmonare idiopatica. Il potenziale impatto dell'inibizione del VEGFR sulla patologia IPF non è del tutto chiarito al momento. Si ipotizza che nintedanib inibisca a livello molecolare le cascate di segnali FGFR e PDGFR, che mediano la proliferazione e la migrazione dei fibroblasti polmonari, legandosi al sito di legame per l'adenosina trifosfato (ATP) del dominio chinasico recettoriale intracellulare e altera in tal modo l'attivazione crociata (tramite autofosforilazione degli omodimeri dei recettori).
In vitro, nintedanib inibisce i suoi recettori bersaglio a basse concentrazioni nanomolari. Nei pazienti con IPF, nintedanib ha inibito la proliferazione dei fibroblasti polmonari stimolata da PDGF, FGF e VEGF con valori di EC50 pari a 11 nmol/l, 5,5 nmol/l o inferiori a 1 nmol/l. A concentrazioni tra 100 e 1.000 nmol/l, nintedanib ha inibito inoltre la migrazione dei fibroblasti stimolata da PDGF, FGF e VEGF e la trasformazione dei fibroblasti in miofibroblasti indotta da TGF-β2. Si ipotizza inoltre che l'attività antinfiammatoria di nintedanib limiti la stimolazione fibrotica riducendo i mediatori profibrotici come IL-1β e IL-6. Non è stato ancora chiarito in che misura l'attività anti-angiogenica di nintedanib concorra al meccanismo d'azione del medicamento sulle patologie polmonari fibrotiche. Negli studi in vivo nintedanib ha prodotto una marcata attività antifibrotica e antinfiammatoria.
Efficacia clinica
Fibrosi polmonare idiopatica (IPF)
L'efficacia clinica di nintedanib è stata studiata in uno studio di fase II (TOMORROW) e in due studi di fase III (INPULSIS-1 e INPULSIS-2) in 1231 pazienti con IPF. Questi studi randomizzati, controllati con placebo, in doppio cieco, hanno confrontato un trattamento con Ofev 150 mg due volte al giorno e con placebo per la durata di 52 settimane.
Gli studi INPULSIS-1 e INPULSIS-2 avevano lo stesso disegno, mentre lo studio TOMORROW aveva un disegno molto simile. I pazienti sono stati randomizzati con un rapporto di 3:2 (nello studio TOMORROW 1:1) al trattamento con Ofev 150 mg o placebo, rispettivamente due volte al giorno per 52 settimane. Lo studio TOMORROW presentava inoltre altri bracci di trattamento (50 mg al giorno, 50 mg due volte al giorno e 100 mg due volte al giorno), che non vengono approfonditi in questa sede.
L'endpoint primario era il tasso annuale di declino della capacità vitale forzata (CVF). La variazione del punteggio totale del Saint George's Respiratory Questionnaire (SGRQ) tra l'inizio dello studio e la settimana 52 e il tempo alla prima esacerbazione acuta dell'IPF erano gli endpoint secondari principali degli studi INPULSIS e gli endpoint secondari dello studio TOMORROW.
Tasso annuale di declino della CVF
Il tasso annuale di declino della CVF (in ml) risultava significativamente ridotto nei pazienti trattati con nintedanib rispetto ai pazienti trattati con placebo. L'effetto del trattamento è risultato coerente in tutti e 3 gli studi. La Tabella 1 mostra i risultati per i singoli studi e i risultati aggregati per l'analisi INPULSIS.
Tabella 1: Tasso annuale di declino della CVF (ml) negli studi TOMORROW, INPULSIS-1 e INPULSIS-2 e dati aggregati relativi all'analisi INPULSIS – popolazione trattata2
TOMORROW | INPULSIS-1 | INPULSIS-2 | INPULSIS-1 e INPULSIS-2 Dati aggregati | |||||
|---|---|---|---|---|---|---|---|---|
Placebo | Ofev 150 mg due volte al giorno | Placebo | Ofev 150 mg due volte al giorno | Placebo | Ofev 150 mg due volte al giorno | Placebo | Ofev 150 mg due volte al giorno | |
Numero di pazienti analizzati | 83 | 84 | 204 | 309 | 219 | 329 | 423 | 638 |
Tasso 1,2 (ES) di declino in 52 settimane | -190 (36) | -60 (39) | -239,9 (18,71) | -114,7 (15,33) | -207,3 (19,31) | -113,6 (15,73) | -223,5 (13,45) | -113,6(10,98) |
Confronto rispetto al placebo | ||||||||
Differenza1 | 131 | 125,3 | 93,7 | 109,9 | ||||
IC al 95% | (27; 235) | (77,7; 172,8) | (44,8; 142,7) | (75,9; 144,0) | ||||
Valore p | 0,01363 | <0,0001 | 0,0002 | <0,0001 | ||||
1 Calcolo effettuato sulla base di un modello di regressione a coefficienti casuali. 2 Popolazione randomizzata nello studio TOMORROW; popolazione trattata negli studi INPULSIS-1, INPULSIS-2 e nei dati aggregati dell'analisi INPULSIS 3 Valore p nominale. | ||||||||
La robustezza dell'effetto di nintedanib sulla riduzione del tasso annuale di declino della CVF è stata confermata in tutte le analisi di sensibilità pre-specificate. Inoltre, sono stati osservati effetti simili su altri endpoint relativi alla funzionalità polmonare, ad esempio la variazione rispetto al basale della CVF alla settimana 52 e l'analisi dei CVF responder, fornendo ulteriori conferme degli effetti di nintedanib sul rallentamento della progressione della malattia. La Figura 1 mostra l'andamento nel tempo della variazione della CVF rispetto al basale in entrambi i gruppi di trattamento (in base all'analisi dei dati aggregati degli studi INPULSIS-1 e INPULSIS-2).
Figura 1: Variazione media (ESM) della CVF rispetto al basale osservata nel tempo (ml); studi INPULSIS-1 e INPULSIS-2 (dati aggregati)

bid = due volte al giorno, ESM = errore standard della media
Variazione rispetto al basale del punteggio totale SGRQ alla settimana 52
Il punteggio totale del Saint George's Respiratory Questionnaire (SGRQ), che misura la qualità della vita correlata alla salute (HRQoL), è stato analizzato a 52 settimane. Nello studio TOMORROW, la variazione media stimata del punteggio totale SGRQ tra l'inizio dello studio (basale) e la settimana 52 nel gruppo placebo era 5,46, indicando un peggioramento della qualità della vita correlata alla salute, mentre nel gruppo nintedanib era -0,66, confermando una qualità della vita correlata alla salute stabile. La differenza media stimata tra nintedanib e placebo era -6,12 (IC al 95%: -10,57; -1,67; p = 0,0071).
Nello studio INPULSIS-2, i pazienti trattati con placebo presentavano un maggiore aumento rispetto al basale del punteggio totale SGRQ a confronto con i pazienti trattati con nintedanib 150 mg due volte al giorno. Il peggioramento della HRQoL era inferiore nel gruppo nintedanib; la differenza fra i gruppi di trattamento era statisticamente significativa (-2,69; IC al 95%: -4,95; -0,43; p = 0,0197).
Nello studio INPULSIS-1, l'aumento rispetto al basale del punteggio totale del SGRQ alla settimana 52 era di entità paragonabile fra il gruppo nintedanib e il gruppo placebo (differenza fra i gruppi di trattamento: -0,05; IC al 95%: -2,50; 2,40; p = 0,9657). Nell'analisi dei dati aggregati degli studi INPULSIS, la variazione media stimata del punteggio totale SGRQ tra il basale e la settimana 52 era inferiore nel gruppo nintedanib (3,53) rispetto al gruppo placebo (4,96), con una differenza fra i gruppi di trattamento di -1,43 (IC al 95%: -3,09; 0,23; p = 0,0923). Complessivamente, l'effetto di nintedanib sulla qualità della vita correlata alla salute misurato tramite il punteggio totale SGRQ è modesto e indica un peggioramento inferiore rispetto al trattamento con placebo.
Tempo alla prima esacerbazione acuta dell'IPF
Negli studi TOMORROW e INPULSIS-2, il rischio della prima esacerbazione acuta dell'IPF nel periodo di 52 settimane è risultato significativamente ridotto nei pazienti trattati con nintedanib rispetto a quelli trattati con placebo (Hazard Ratio [HR]: 0,16; IC al 95%: 0,04; 0,71; p = 0,0054) o (HR: 0,38; IC al 95%: 0,19; 0,77; p = 0,005). Nello studio INPULSIS-1 non vi era differenza fra i gruppi di trattamento (HR: 1,15; IC al 95%: 0,54; 2,42; p = 0,6728).
Analisi della sopravvivenza
Nell'analisi pre-specificata dei dati aggregati sulla sopravvivenza degli studi INPULSIS, la mortalità complessiva su 52 settimane è risultata numericamente inferiore nel gruppo nintedanib (5,5%) rispetto al gruppo placebo (7,8%). L'analisi del tempo al decesso ha prodotto un HR di 0,70 (IC al 95%: 0,43; 1,12; p = 0,1399). I risultati di tutti gli endpoint relativi alla sopravvivenza (quali la mortalità in trattamento e la mortalità respiratoria) hanno mostrato una costante differenza numerica a favore di nintedanib, senza tuttavia raggiungere una significanza statistica.
Ulteriori dati dello studio INJOURNEY di fase IV con Ofev 150 mg due volte al giorno in associazione con pirfenidone:
Uno studio esplorativo, randomizzato, in aperto ha valutato la co-somministrazione di nintedanib 150 mg due volte al giorno con pirfenidone (titolato a 801 mg tre volte al giorno) rispetto a nintedanib 150 mg due volte al giorno in monoterapia in 105 pazienti per la durata di 12 settimane. L'endpoint primario era la percentuale di pazienti con eventi avversi gastrointestinali dall'inizio dello studio alla settimana 12. Gli eventi avversi gastrointestinali sono stati frequenti e in linea con il profilo di sicurezza definito per ciascuna delle due sostanze. Diarrea, nausea e vomito sono stati gli eventi avversi più frequenti, segnalati in 20 (37,7%) pazienti rispetto a 16 (31,4%), 22 (41,5%) pazienti rispetto a 6 (11,8%) o 15 (28,3%) pazienti rispetto a 6 (11,8%) rispettivamente dei pazienti trattati con pirfenidone in aggiunta a nintedanib e di quelli trattati con nintedanib in monoterapia.
Le variazioni assolute medie (ES) della CVF dal basale alla settimana 12 erano −13,3 (17,4) mL nei pazienti trattati con pirfenidone in aggiunta a nintedanib (n=48) rispetto a −40,9 (31,4) mL nei pazienti trattati con nintedanib in monoterapia (n=44).
Malattie interstiziali polmonari (ILD) fibrosanti croniche con fenotipo progressivo
L'efficacia clinica di OFEV nei pazienti con ILD fibrosanti croniche con fenotipo progressivo è stata esaminata in uno studio di fase III, randomizzato, in doppio cieco, controllato con placebo (INBUILD).
Sono stati randomizzati 1:1 complessivamente 663 pazienti, che hanno ricevuto OFEV 150 mg due volte al giorno oppure placebo per almeno 52 settimane.
La randomizzazione è stata stratificata secondo il pattern fibrotico osservato alla tomografia computerizzata ad alta risoluzione (HRCT). Sono stati randomizzati 412 pazienti con pattern fibrotico UIP-simile (usual interstitial pneumonia, polmonite interstiziale usuale) e 251 pazienti con altri pattern fibrotici. Per l'analisi dello studio sono state definite 2 popolazioni co-primarie: la popolazione totale e la popolazione con pattern fibrotico UIP-simile osservato alla HRCT.
L'endpoint primario era il tasso annuale di declino della CVF (ml) in 52 settimane. Gli endpoint dello studio erano il tempo alla prima esacerbazione acuta dell'ILD o al decesso oppure il tempo al decesso.
I pazienti con diagnosi clinica di ILD fibrosante cronica sono stati selezionati per lo studio se presentavano una fibrosi rilevante alla HRCT (>10% di caratteristiche fibrotiche) e segni clinici di progressione della malattia (definita come declino della CVF ≥10%, declino della CVF tra 5 e 10% con peggioramento dei sintomi della malattia O del quadro radiologico oppure con peggioramento dei sintomi della malattia E peggioramento del quadro radiologico) nei 24 mesi precedenti nonostante un adeguato trattamento. La CVF doveva essere ≥45% del valore previsto e la DLCO doveva essere tra il 30% e l'80% del valore previsto.
Sono stati esclusi dalla partecipazione allo studio i pazienti con
- IPF, broncocostrizione rilevante (ad es. FEV1/CVF <0,7 prima della broncodilatazione) oppure ipertensione polmonare significativa
- aumento delle transaminasi o della bilirubina >1,5 ULN
- noto rischio di sanguinamento, trattamento con dose intera di anticoagulante, recente infarto miocardico o ictus
- precedente trattamento con nintedanib, pirfenidone, azatioprina, ciclosporina, micofenolato, tacrolimus, coticosteroidi orali a dosi >20 mg/giorno o coticosteroidi orali + azatioprina + N-acetilcisteina nelle 4 settimane precedenti la randomizzazione*
- precedente trattamento con ciclofosfamide nelle 8 settimane precedenti la randomizzazione*
- precedente trattamento con rituximab nei 6 mesi precedenti la randomizzazione*
* l'impiego dei suddetti medicamenti era consentito nuovamente 6 mesi dopo l'inizio dello studio, se clinicamente indicato
I partecipanti allo studio erano prevalentemente di etnia caucasica (74%) e asiatica (25%). Il 54% dei pazienti era di sesso maschile, il 49% non aveva mai fumato, l'età media era di 66 anni e la CVF era in media pari al 69% del valore previsto. Le ILD di base erano polmonite da ipersensibilità (26%), ILD autoimmuni (26%), polmonite interstiziale idiopatica aspecifica (19%), polmonite interstiziale idiopatica non classificabile (17%) e altre ILD (12%).
Tasso annuale di declino della CVF (ml)
In 52 settimane è stata osservata una riduzione statisticamente significativa del declino della CVF nei pazienti trattati con OFEV rispetto a quelli trattati con placebo. La differenza tra OFEV e placebo è stata di 107 ml (Tabella 2).
Tabella 2: Tasso annuale di declino della CVF (ml, endpoint primario dello studio INBUILD)
Popolazione totale | Sottopopolazione pattern UIP-simile | Sottopopolazione Altri pattern fibrotici alla HRCT | ||||
|---|---|---|---|---|---|---|
OFEV | Placebo | OFEV | Placebo | OFEV | Placebo | |
Numero di pazienti analizzati | 332 | 331 | 206 | 206 | 126 | 125 |
Tassoa (ES) di declino in 52 settimane | -80,8 | -187,8 | -82,9 | -211,1 | -79,0 | -154,2 |
Confronto verso placebo | 107,0 | 128,2 | 75,2* | |||
IC 95% | (65,4, 148,5) | (70,8, 185,6) | (15,5, 135,0)* | |||
Valore p | < 0,0001 | < 0,0001 | ||||
* Nella procedura di analisi multipla non è stato incluso un confronto basato sulla sottopopolazione con altri pattern fibrotici alla HRCT. I valori qui forniti hanno finalità descrittiva. a Basato su una regressione a coefficienti casuali con effetti categorici fissi di trattamento, pattern alla HRCT, effetti continui fissi di tempo, CVF [ml] basale e comprendendo le interazioni trattamento-tempo e basale-tempo. | ||||||
La Figura 2 mostra la variazione nel tempo della CVF rispetto al basale nei gruppi di trattamento.
Figura 2: Variazione media (ESM) della CVF (ml) rispetto al basale osservata in 52 settimane
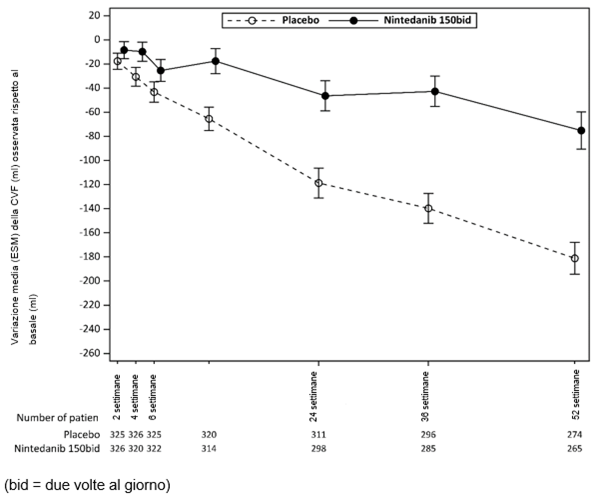
È stata effettuata un'analisi esplorativa post-hoc dopo la diagnosi di ILD (Figura 3). Complessivamente, è stato evidenziato un effetto terapeutico coerente nelle diverse diagnosi di ILD.
Figura 3: Tasso annuale di declino della CVF (ml) in 52 settimane in base alla diagnosi di ILD di base nello studio 5*
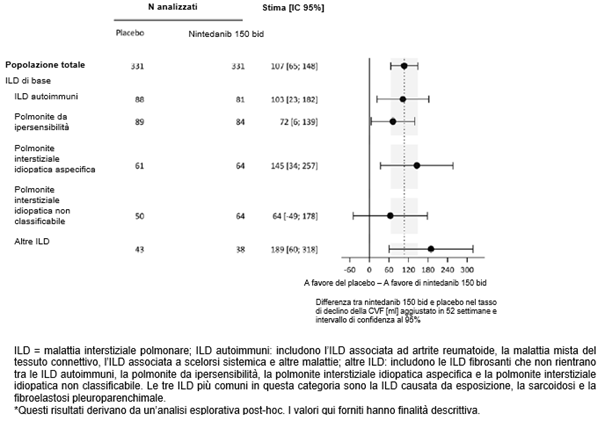
Declino percentuale della CVF
La Figura 4 mostra la variazione percentuale della CVF (ml) dal basale alla settimana 52. Per la maggior parte dei pazienti, il declino della funzionalità polmonare in terapia con OFEV è stato inferiore rispetto alla terapia con placebo.
Figura 4: Istogramma del declino percentuale della CVF (ml) rispetto al basale alla settimana 52 dopo trattamento, con incremento o riduzione percentuale a passi di 5 (Studio 5)a
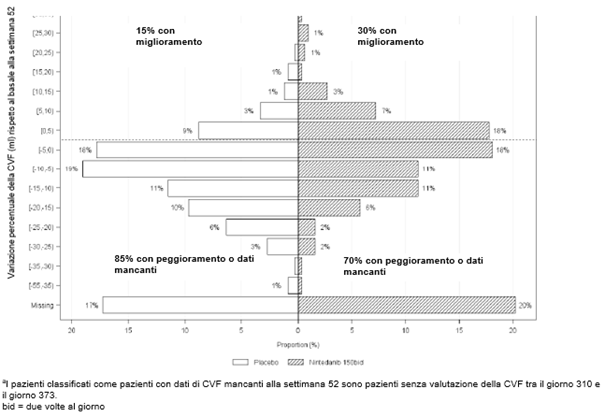
Tempo alla prima esacerbazione acuta dell'ILD o al decesso
Il rischio di prima esacerbazione acuta dell'ILD o di decesso si è ridotto nel gruppo Ofev rispetto al gruppo placebo in 52 settimane (HR Ofev versus placebo: 0,80; IC 95%: 0,48; 1,34) o nel corso dell'intera durata dello studio (HR Ofev versus placebo: 0,67; IC 95%: 0,46; 0,98), indicando una riduzione del 33% del rischio di prima esacerbazione acuta dell'ILD o di decesso nei pazienti trattati con Ofev rispetto al placebo.
Analisi della sopravvivenza
Nello studio INBUILD i dati di sopravvivenza in terapia con Ofev sono stati confrontati con quelli in terapia con placebo per avvalorare l'endpoint primario (CVF). La mortalità per tutte le cause è stata valutata per l'intera durata dello studio e per il periodo di follow-up disponibile, indipendentemente dalla causa di morte e dal proseguimento o meno del trattamento. L'analisi ha evidenziato una differenza non statisticamente significativa in 52 settimane (HR OFEV versus placebo 0,94, IC 95% 0,47, 1,86) e/o per l'intera durata dello studio (HR OFEV versus placebo 0,78, IC 95% 0,5, 1,21).
Malattia interstiziale polmonare associata a sclerosi sistemica (SSc-ILD)
L'efficacia clinica di nintedanib è stata esaminata su pazienti con SSc-ILD in uno studio di fase III (SENSCIS), randomizzato, in doppio cieco, controllato verso placebo.
Complessivamente, 580 pazienti sono stati randomizzati in un rapporto 1:1 per ricevere Ofev 150 mg due volte al giorno o placebo per almeno 52 settimane. La randomizzazione è stata stratificata in base allo stato degli anticorpi anti-topoisomerasi (ATA). Alcuni pazienti hanno proseguito il trattamento di studio in cieco per un periodo di fino a 100 settimane.
La diagnosi dei pazienti con SSc-ILD si basava sui criteri di classificazione per la SSc dell'American College of Rheumatology / della European League Against Rheumatism, se l'insorgenza della malattia (primi sintomi non riconducibili al fenomeno di Raynaud) risaliva a meno di 7 anni prima e se era presente almeno una fibrosi polmonare del 10%, ed era stata accertata mediante una tomografia computerizzata ad alta risoluzione (HRCT) eseguita nei 12 mesi precedenti.
La CVF nei pazienti doveva essere pari almeno al 40% del valore stimato, mentre la DLCO doveva essere pari al 30‑89% del valore stimato. I pazienti con ostruzioni rilevanti delle vie aeree (ovvero FEV1/CVF prima della broncodilatazione inferiore a 0,7) oppure sottoposti o in procinto di sottoporsi ad un trapianto di cellule staminali ematopoietiche non hanno potuto partecipare allo studio. Altri criteri di esclusione sono stati un aumento dei valori di ALT, AST o bilirubina (>1,5 x ULN), un aumento del rischio di sanguinamenti (inclusa terapia anticoagulante), anamnesi di recente infarto miocardico o ictus, significativa ipertensione polmonare, ulcere ad almeno tre polpastrelli delle dita, anamnesi di grave necrosi digitale con necessaria ospedalizzazione e anamnesi di crisi renale sclerodermica. Erano esclusi anche i pazienti che avevano ricevuto altri medicamenti sperimentali, che erano già stati trattati con nintedanib o pirfenidone e che avevano ricevuto azatioprina 8 settimane prima della randomizzazione oppure ciclofosfamide o ciclosporina A 6 mesi prima della randomizzazione. Era consentita una terapia stabile con micofenolato o metotressato e con prednisone ≤10 mg/die o equivalente.
L'endpoint primario era il tasso annuale di declino della capacità vitale forzata (CVF) in 52 settimane.
Gli endpoint secondari importanti erano la variazione assoluta rispetto al basale del punteggio Rodnan Skin Score modificato (mRSS) alla settimana 52, la variazione assoluta rispetto al basale del punteggio totale Saint George's Respiratory Questionnaire (SGRQ) alla settimana 52 e il tasso di mortalità durante l'intero studio.
Il 75,2% dei pazienti era di sesso femminile. I pazienti erano prevalentemente di etnia caucasica (67%), asiatica (25%) o nera (6%). L'età media (deviazione standard [DS], min.-max.) era di 54,0 (12,2, 20–79) anni. Complessivamente, il 51,9% dei pazienti era affetto da sclerosi sistema cutanea diffusa (SSc) e il 48,1% da SSc cutanea limitata. Il tempo medio (DS) dalla prima insorgenza di sintomi non riconducibili al fenomeno di Raynaud era di 3,49 (1,7) anni. Il 49,0% dei pazienti dei pazienti era stabilmente in terapia con micofenolato al basale e il profilo di sicurezza dei pazienti con o senza micofenolato al basale era comparabile.
Tasso annuale di declino della CVF
Il tasso annuale di declino della CVF (in ml) in 52 settimane si è significativamente ridotto di 41,0 ml nei pazienti trattati con Ofev rispetto a quelli trattati con placebo (vedere Tabella 3), corrispondente a un effetto di trattamento relativo del 43,8%.
Tabella 3: Tasso annuale di declino della CVF (ml) in 52 settimane
Placebo | Ofev 150 mg due volte al giorno | |
|---|---|---|
Numero di pazienti analizzati | 288 | 287 |
Tasso1 (ES) di declino in 52 settimane | -93,3 (13,5) | -52,4 (13,8) |
Confronto verso placebo | ||
Differenza1 | 41,0 | |
IC 95% | (2,9, 79,0) | |
Valore p | <0,05 | |
1 Basato su una regressione a coefficienti casuali, aggiustati per trattamento, sesso, altezza, età, stato ATA, CVF basale, interazioni CVF-tempo e trattamento-tempo | ||
Un'analisi esplorativa dei dati fino a 100 settimane (durata massima del trattamento nello studio SENSCIS) ha indicato che l'effetto del trattamento con Ofev sul rallentamento della progressione della SSc-ILD è continuato oltre le 52 settimane.
In due analisi predefinite per sottogruppi relative all'efficacia è stata studiata la differenza media del trattamento in relazione al tasso di declino della CVF alla settimana 52 nei pazienti in base alla regione geografica e all'uso di micofenolato (vedere Figura 5).
Figura 5: Analisi per sottogruppi della differenza media del trattamento in relazione al tasso di declino della CVF (ml) alla settimana 52 in base alla regione geografica e all'uso di micofenolato (SENSCIS)
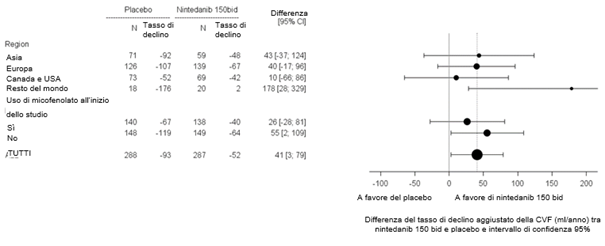
Variazione della percentuale rispetto al basale della capacità vitale forzata
La Figura 6 illustra per lo studio SENSCIS la variazione percentuale della CVF in ml rispetto al basale alla settimana 52. Il peggioramento della funzionalità polmonare era meno marcato nella maggior parte dei pazienti trattati con Ofev rispetto al placebo.
Figura 6: Istogramma della variazione percentuale della CVF (ml) rispetto al basale alla settimana 52 in base al trattamento e all'incremento o alla riduzione percentuale di 5 (SENSCIS)a
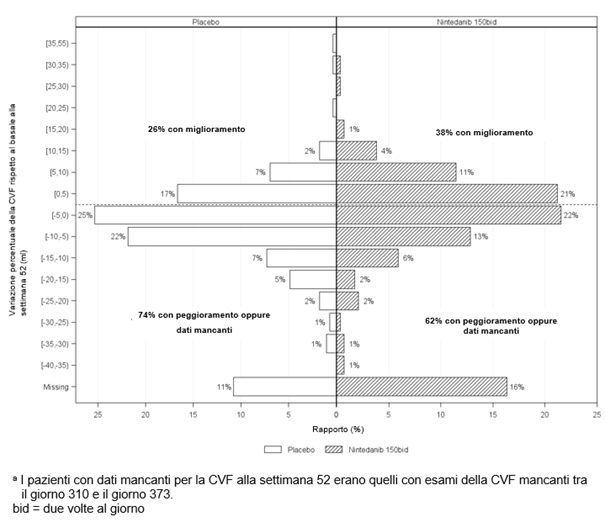
Figura 7: Variazione media (ESM) della CVF rispetto al basale (ml) osservata in 52 settimane

Tabella 4 Tasso annuale di declino della CVF (% prevista) in 52 settimane
Placebo | Ofev 150 mg due volte al giorno | |
|---|---|---|
Numero di pazienti analizzati | 288 | 287 |
Tasso1 (ES) di declino in 52 settimane | -2,6 (0,4) | -1,4 (0,4) |
Confronto verso placebo | ||
Differenza1 | 1,15 | |
IC 95% | (0,09, 2,21) | |
Valore p | <0,05 | |
1 Basato su una regressione a coefficienti casuali ed effetti categorici fissi di trattamento, stato ATA, effetti continui fissi di tempo, CVF basale [% prevista] e comprendendo le interazioni trattamento-tempo e basale-tempo. Sono stati inclusi gli effetti casuali di intercetta e tempo a livello del paziente. Gli errori intrinseci al paziente (within-patient error) sono stati modellati secondo una matrice di varianza e covarianza non strutturata. La variabilità interindividuale è stata modellata secondo una matrice di varianza e covarianza a componenti di varianza. | ||
Variazione rispetto al basale del punteggio Rodnan Skin Score modificato (mRSS) alla settimana 52
La variazione assoluta della media aggiustata rispetto al basale del punteggio mRSS alla settimana 52 era comparabile tra il gruppo Ofev (-2,17 (IC 95% -2,69, -1,65)) e il gruppo placebo (-1,96 (IC 95% -2,48, -1,45)). La differenza media aggiustata tra i gruppi di trattamento era -0,21 (IC 95% -0,94, 0,53; p = 0,5785).
Variazione rispetto al basale del punteggio totale St. George's Respiratory Questionnaire (SGRQ) alla settimana 52
La variazione assoluta della media aggiustata rispetto al basale del punteggio totale SGRQ alla settimana 52 era comparabile tra il gruppo Ofev (0,81 (IC 95% -0,92, 2,55)) e il gruppo placebo (-0,88 (IC 95% -2,58, 0,82)). La differenza media aggiustata tra i gruppi di trattamento era 1,69 (IC 95% -0,73, 4,12; p = 0,1711).
Analisi della sopravvivenza
La mortalità complessiva nell'intero studio era comparabile tra il gruppo Ofev (N = 10; 3,5%) e il gruppo placebo (N = 9; 3,1%). L'analisi del tempo al decesso nell'intero studio ha prodotto un HR di 1,16 (IC 95% 0,47, 2,84; p = 0,7535).
Effetto sull'intervallo QT
In uno studio speciale su pazienti con carcinoma a cellule renali sono state registrate le misure degli intervalli QT/QTc ed è stato mostrato che una dose orale singola di 200 mg di nintedanib o dosi orali multiple di 200 mg di nintedanib somministrate due volte al giorno per 15 giorni non hanno prolungato l'intervallo QTcF. Non è stato condotto uno studio clinico completo sul QT (Thorough-QT-Study) con nintedanib.
Studi su bambini e adolescenti
Non sono stati condotti studi clinici su bambini e adolescenti.
Farmacocinetica
Le proprietà farmacocinetiche di nintedanib erano simili nei soggetti di studio sani, nei pazienti con IPF, SSc-ILD, ILD e nei pazienti oncologici.
La farmacocinetica di nintedanib è lineare rispetto al tempo. La proporzionalità della dose era dimostrata dall'aumento dell'esposizione a nintedanib (intervallo della dose 50–450 mg una volta al giorno e 150–300 mg due volte al giorno). L'accumulo dopo somministrazioni multiple a pazienti con IPF era pari a 1,76 volte per l'AUCτ. Le concentrazioni plasmatiche allo stato stazionario sono state raggiunte dopo 1 settimana. Le concentrazioni minime di nintedanib sono rimaste stabili per oltre un anno. La farmacocinetica di nintedanib presenta una variabilità interindividuale moderata-elevata (coefficiente di variazione per parametri farmacocinetici standard nell'intervallo dal 30% al 70%). La variabilità intraindividuale è bassa-moderata (coefficiente di variazione inferiore al 40%).
Assorbimento
Le concentrazioni plasmatiche massime di nintedanib sono state raggiunte circa 2–4 ore dopo l'assunzione orale di capsule molli di gelatina a stomaco pieno (intervallo: 0,5–8 ore). La biodisponibilità assoluta di una dose di 100 mg era pari al 4,69% nei soggetti di studio sani (IC al 90%: 3,62–6,08). L'assorbimento e la biodisponibilità vengono ridotti dagli effetti di trasportatori (P-gp), da un sostanziale metabolismo di primo passaggio ed eventualmente dalla scarsa solubilità di nintedanib in condizioni di pH neutro.
Dopo l'assunzione di cibo l'esposizione a nintedanib aumentava di circa il 20% rispetto alla somministrazione in condizioni di digiuno (IC: 95,3–152,5%) e l'assorbimento risultava rallentato (tmax mediano a digiuno: 2,00 ore; dopo l'assunzione di cibo: 3,98 ore).
Distribuzione
Nintedanib segue una cinetica di distribuzione almeno bifasica. Dopo l'infusione endovenosa è stato osservato un volume elevato di distribuzione (Vss: 1050 litri).
Il legame proteico in vitro di nintedanib nel plasma umano era elevato (97,8%). L'albumina sierica è considerata la proteina con maggiore capacità di legame. Nintedanib è distribuito principalmente nel plasma, con un rapporto sangue/plasma pari a 0,869.
Metabolismo
La via metabolica principale del nintedanib è la scissione idrolitica tramite le esterasi, che porta alla formazione di BIBF 1202 con il gruppo funzionale acido libero. BIBF 1202 viene successivamente trasformato dagli enzimi UGT 1A1, UGT 1A7, UGT 1A8 e UGT 1A10 in BIBF 1202 glucuronide.
Solo una quantità ridotta della biotrasformazione di nintedanib avviene tramite gli enzimi del CYP, prevalentemente CYP 3A4. Nello studio ADME la percentuale principale di radioattività nel plasma umano è stata attribuita a nintedanib (24%), a BIBF 1202 (32%) e a BIBF 1202 glucuronide (30%). Non è stato possibile rilevare i principali metaboliti CYP-dipendenti nel plasma umano nello studio ADME. In vitro, il metabolismo CYP-dipendente era responsabile di circa il 5%, rispetto al 25% circa della scissione degli esteri nelle condizioni sperimentali indicate.
Eliminazione
La clearance plasmatica totale dopo l'infusione endovenosa era elevata (CL: 1390 ml/min). Entro 48 ore dalla somministrazione, l'escrezione urinaria del principio attivo immodificato era pari allo 0,05% della dose in caso di somministrazione orale e a circa l'1,4% in caso di somministrazione endovenosa. La clearance renale era pari a 20 ml/min.
Escrezione
La principale via di eliminazione della radioattività correlata al medicamento dopo somministrazione orale di [14C]-nintedanib era l'escrezione per via fecale/biliare (93,4% della dose). La maggior parte di Ofev veniva escreta sotto forma di BIBF 1202 tramite le feci (58% della dose); il 20% veniva escreto come sostanza madre e meno del 7% sotto forma di vari metaboliti meno importanti. Il contributo all'escrezione renale della clearance totale era basso (0,649% della dose). Il recupero era considerato completo (oltre il 90%) entro i 4 giorni successivi alla somministrazione. L'emivita terminale di nintedanib era tra 10 e 15 ore.
Rapporto esposizione/risposta
Le analisi esplorative riguardanti il rapporto tra farmacocinetica ed eventi avversi eseguite sui dati dello studio di fase II sull'IPF hanno mostrato che una maggiore esposizione a nintedanib è tendenzialmente correlata ad un aumento degli enzimi epatici (vedere la rubrica «Avvertenze e misure precauzionali»).
Cinetica di gruppi di pazienti speciali, fattori intrinseci ed estrinseci
In base ai risultati di un'analisi di farmacocinetica di popolazione condotta su pazienti con IPF e carcinoma polmonare non a piccole cellule (NSCLC) (N = 1191) e in base a ricerche descrittive, l'esposizione a nintedanib non è stata influenzata da fattori quali il sesso (corretto in base al peso corporeo), la compromissione renale lieve e moderata (stimata in base alla clearance della creatinina), il consumo di alcol e il genotipo P-gp. L'analisi di farmacocinetica di popolazione ha indicato effetti modesti sull'esposizione a nintedanib dei fattori età, peso corporeo ed etnia descritti qui di seguito. In base all'elevata variabilità interindividuale, gli effetti modesti di queste covariabili non giustificano un aggiustamento della dose (vedere la rubrica «Avvertenze e misure precauzionali»).
Età
L'esposizione a nintedanib aumenta in modo lineare con l'età. L'AUCτ,ss era diminuita del 16% per un paziente di 45 anni (5° percentile) ed era aumentata del 13% per un paziente di 76 anni (95° percentile) rispetto ad un paziente di 62 anni (età mediana). L'analisi copriva un intervallo di età dai 29 agli 85 anni e circa il 5% della popolazione aveva più di 75 anni.
Non sono stati condotti studi su bambini e adolescenti.
Peso corporeo
È stata osservata una correlazione inversa fra peso corporeo ed esposizione a nintedanib. L'AUCτ,ss era aumentata del 25% per un paziente di 50 kg (5° percentile) ed era diminuita del 19% per un paziente di 100 kg (95° percentile) rispetto ad un paziente di 71,5 kg (peso mediano).
Etnia
L'esposizione media della popolazione a nintedanib era maggiore del 33–50% nei pazienti cinesi, taiwanesi e indiani ed era maggiore del 16% nei pazienti giapponesi, mentre era inferiore del 16–22% nei pazienti coreani rispetto ai pazienti caucasici (dati corretti in base al peso corporeo). I dati disponibili sui soggetti di etnia nera erano molto limitati, ma rientravano nello stesso range dei dati disponibili per i soggetti caucasici.
Fumatori
L'analisi farmacocinetica di popolazione ha evidenziato che l'esposizione a nintedanib nei fumatori attivi era inferiore del 21% rispetto a quella negli ex fumatori e nei non fumatori per tutta la vita. Un effetto di questa portata non giustifica un aggiustamento della dose.
Disturbi della funzionalità epatica
In uno specifico studio di fase I su dose singola confrontato con soggetti sani, l'esposizione a nintedanib in base alla Cmax e all'AUC era 2,2 volte maggiore nei volontari con compromissione epatica lieve (classe Child-Pugh A; IC al 90% 1,3–3,7 per Cmax o 1,2–3,8 per AUC). Nei volontari con compromissione epatica moderata (classe Child-Pugh B), l'esposizione era 7,6 volte maggiore in base alla Cmax (IC al 90% 4,4–13,2) e 8,7 volte maggiore (IC al 90% 5,7–13,1) in base all'AUC, rispetto ai volontari sani. I soggetti con compromissione epatica severa (classe Child Pugh C) non sono stati studiati.
Disturbi della funzionalità renale
La farmacocinetica di nintedanib non è stata studiata in uno studio FC speciale su pazienti con compromissione renale. In base ai risultati dell'analisi di farmacocinetica di popolazione, una compromissione renale lieve (CrCl: 60–90 ml/min) o moderata (CrCl: 30–60 ml/min) non ha influenzato l'esposizione a nintedanib. Sono disponibili solo dati limitati su pazienti con compromissione renale severa (CrCl inferiore a 30 ml/min).
Dati preclinici
Tossicologia generale
Gli studi di tossicità a dose singola nel ratto e nel topo hanno indicato un basso potenziale di tossicità acuta per nintedanib. Negli studi di tossicità a dose ripetuta nel ratto, gli effetti indesiderati (ad esempio ispessimento delle placche epifisarie, lesioni a carico degli incisivi) erano correlati principalmente al meccanismo d'azione di nintedanib (ovvero inibizione del VEGFR-2). Queste variazioni sono note per altri inibitori del VEGFR-2 e possono essere considerate effetti di classe.
In studi di tossicità a dose ripetuta condotti su specie non roditori sono stati osservati diarrea e vomito, accompagnati da riduzione del consumo di cibo e da diminuzione del peso.
Non vi era evidenza di aumento degli enzimi epatici nel ratto, nel cane e nei macachi a coda lunga (scimmie Cynomolgus). Lievi aumenti degli enzimi epatici, non dovuti ad effetti indesiderati severi come la diarrea, sono stati osservati solo nelle scimmie Rhesus.
Mutagenicità/Cancerogenicità
Gli studi di genotossicità non hanno indicato potenziale mutageno per nintedanib.
Studi di cancerogenicità della durata di 2 anni nel topo e nel ratto non hanno prodotto evidenze di potenziale cancerogeno per nintedanib.
Tossicità per la riproduzione
Nel ratto sono stati osservati mortalità embrio-fetale ed effetti teratogeni a livelli di esposizione inferiori di 3,6–7,2 volte rispetto alla dose massima raccomandata nell'uomo (MRHD) di 150 mg due volte al giorno. A livelli di esposizione inferiori di circa 12–18 volte rispetto alla MRHD sono stati riscontrati effetti irrilevanti sullo sviluppo dello scheletro assiale e sullo sviluppo delle grandi arterie.
Nel coniglio sono stati osservati mortalità embrio-fetale ed effetti teratogeni a livelli di esposizione superiori di circa 3 volte rispetto alla MRHD, mentre è stata riscontrata una compromissione meno evidente sullo sviluppo embrio-fetale dello scheletro assiale e del cuore già a livelli di esposizione inferiori alla MRHD di 150 mg due volte al giorno.
Uno studio condotto sulla fertilità maschile e sulle fasi iniziali dello sviluppo embrionale fino all'impianto nel ratto non ha rivelato effetti sul tasso di riproduzione e sulla fertilità maschile a livelli di esposizione superiori di circa 3 volte rispetto alla MRHD.
Nel ratto, piccole quantità di nintedanib radiomarcato e/o dei suoi metaboliti sono state escrete nel latte (≤0,5% della dose somministrata).
Altre indicazioni
Stabilità
Il medicamento non deve essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Indicazioni particolari concernenti l'immagazzinamento
Conservare fuori dalla portata dei bambini.
Conservare a temperatura ambiente (15-25 °C). Conservare il contenitore nella scatola originale per proteggere il contenuto dall'umidità.
Numero dell'omologazione
65330 (Swissmedic)
Titolare dell’omologazione
Boehringer Ingelheim (Schweiz) GmbH, Basilea
Stato dell'informazione
Ottobre 2020
Composition
Principes actifs
Nintédanib (sous forme d'ésilate).
Excipients
- Contenu des capsules: triglycérides à chaîne moyenne, graisse solide, lécithine (de soja) (E322)
- Enveloppe de la capsule: gélatine, glycérol (85 %), dioxyde de titane (E171), oxyde de fer jaune (E172), oxyde de fer rouge (E172)
- Encre d'impression: gomme laque, oxyde de fer noir (E172), propylèneglycol (E1520)
Forme pharmaceutique et quantité de principe actif par unité
Capsules molles de 100 mg et de 150 mg de nintédanib (correspondant à 120,40 mg et à 180,60 mg d'ésilate de nintédanib)
Indications/Possibilités d’emploi
Ofev est indiqué pour le traitement
- de la fibrose pulmonaire idiopathique (FPI)
- des pneumopathies interstitielles diffuses (PID) fibrosantes chroniques avec un phénotype progressif (voir rubrique «Efficacité clinique»)
- de la pneumopathie interstitielle diffuse associée à la sclérodermie systémique (PID-ScS)
Posologie/Mode d’emploi
Le traitement doit être instauré par un médecin expérimenté dans le diagnostic et le traitement des maladies pour lesquelles Ofev est indiqué.
La dose recommandée d'Ofev est de 150 mg deux fois par jour, administrée à environ 12 heures d'intervalle.
Les capsules doivent être avalées intactes avec de l'eau lors d'un repas. Elles ne doivent pas être croquées ou partagées d'une quelconque manière.
Si une dose d'Ofev a été omise, le traitement sera poursuivi à la prochaine heure de prise prévue, à la dose recommandée. L'oubli d'une dose ne doit pas être compensé par l'administration d'une dose supplémentaire. La dose maximale journalière recommandée de 300 mg ne doit pas être dépassée.
Ajustement de la posologie
En cas d'effets indésirables d'Ofev (voir les sections «Mises en garde et précautions» et «Effets indésirables»), le traitement symptomatique (si indiqué) peut être accompagné d'une réduction de la dose ou d'une interruption transitoire de l'administration de nintédanib jusqu'à ce que l'effet indésirable en question ait disparu ou suffisamment régressé pour permettre la poursuite du traitement. Le traitement par Ofev peut être repris avec la dose complète (150 mg deux fois par jour) ou à dose réduite (100 mg deux fois par jour). Si la dose de 100 mg deux fois par jour est mal tolérée par le patient, le traitement par Ofev doit être abandonné.
En cas d'interruption du traitement à cause d'une augmentation des taux de transaminases (ASAT ou ALAT) à plus de 3 fois la limite supérieure de la normale (LSN), le traitement par Ofev peut être repris à dose réduite (100 mg deux fois par jour) après la normalisation des taux de transaminases, puis passer ensuite à la dose entière (150 mg deux fois par jour). Dans le cas d'une augmentation du taux d'ASAT ou d'ALAT à plus de 5 fois la LSN ou dans le cas d'une augmentation à plus de 3 fois la LSN en présence de constats ou symptômes indiquant une atteinte hépatique sévère, le traitement par Ofev doit être arrêté (voir «Mises en garde et précautions» et «Effets indésirables»).
Instructions posologiques particulières
Patients présentant des troubles de la fonction rénale
Moins de 1 % d'une dose unique de nintédanib est excrété par les reins (voir «Pharmacocinétique»). Aucun ajustement de la dose initiale n'est donc nécessaire chez les patients présentant une insuffisance rénale légère à modérée. La sécurité, l'efficacité et la pharmacocinétique du nintédanib n'ont pas été étudiées chez des patients atteints d'insuffisance rénale sévère (ClCr <30 ml/min).
Patients présentant des troubles de la fonction hépatique
Le nintédanib est éliminé essentiellement par voie biliaire et dans les selles (>90 %; voir «Pharmacocinétique»). L'exposition systémique est augmentée chez les patients présentant une insuffisance hépatique (Child Pugh A, Child Pugh B). Chez les patients présentant une insuffisance hépatique légère (Child Pugh A), la dose recommandée d'Ofev est de 100 mg deux fois par jour à un intervalle de 12 heures. Chez les patients présentant une insuffisance hépatique légère (Child Pugh A), une réduction de la dose ou un arrêt du traitement doivent être envisagés afin de pouvoir traiter les réactions indésirables.
La sécurité et l'efficacité du nintédanib n'ont pas été étudiées chez les patients présentant une insuffisance hépatique de grade Child-Pugh B ou C. Le traitement par Ofev n'est pas recommandé chez les patients atteints d'une insuffisance hépatique modérée (Child-Pugh B) ou sévère (Child-Pugh C) (voir les sections «Mises en garde et précautions» et «Pharmacocinétique»).
Enfants et adolescents
La sécurité et l'efficacité d'Ofev n'ont pas été examinées dans des études cliniques chez les enfants et adolescents.
Patients âgés (≥65 ans)
Aucune différence concernant la sécurité générale ou l'efficacité n'a été observée entre les patients âgés et ceux de moins de 65 ans. Aucun ajustement posologique n'est nécessaire en fonction de l'âge du patient (voir «Pharmacocinétique»).
Contre-indications
Ofev est contre-indiqué chez les patients présentant une hypersensibilité connue au nintédanib, aux arachides, au soja ou à l'un des autres composants.
Ofev est contre-indiqué pendant la grossesse (voir «Grossesse/Allaitement» et «Données précliniques»).
Mises en garde et précautions
Affections gastro-intestinales
Diarrhée
Dans les études cliniques (voir «Efficacité clinique»), la diarrhée était l'effet indésirable gastro-intestinal le plus fréquemment rapporté.
Dans la majorité des cas, la diarrhée s'est manifestée, avec une intensité légère à modérée dans les 3 premiers mois du traitement.
Dans les études INPULSIS (patients FPI), la diarrhée a été rapportée chez 62,4% des patients sous Ofev contre 18,4% des patients sous placebo. Elle a entraîné une réduction de la dose d'Ofev chez 10,7% des patients contre aucun des patients sous placebo et l'arrêt du traitement par Ofev chez 4,4% des patients contre 0,2% des patients sous placebo.
Dans l'étude INBUILD (patients PID fibrosantes progressives), la diarrhée a été rapportée chez 66,9% des patients sous Ofev contre 23,9% des patients sous placebo. La diarrhée a entraîné une réduction de la dose d'Ofev chez 16,0% des patients contre 0,9% chez les patients sous placebo et l'arrêt du traitement par Ofev chez 5,7% des patients contre 0,3% chez les patients sous placebo.
Dans l'étude SENSCIS (patients PID-ScS), la diarrhée a été rapportée chez 75,7% des patients sous Ofev contre 31,6% des patients sous placebo.
La diarrhée a entraîné une réduction de la dose d'Ofev chez 22,2% des patients contre 1,0% chez les patients sous placebo et l'arrêt du traitement par Ofev chez 6,9% des patients contre 0,3% chez les patients sous placebo (voir rubrique «Effets indésirables»).
La diarrhée peut entraîner une déshydratation avec ou sans troubles électrolytiques, ce qui peut conduire à un dysfonctionnement rénal.
Une diarrhée doit être traitée dès les premiers signes par une réhydratation appropriée et une administration d'antidiarrhéiques tels que le lopéramide. Une réduction de la dose ou l'interruption du traitement est éventuellement nécessaire. Le traitement par Ofev peut être repris à dose réduite (100 mg deux fois par jour) ou entière (150 mg deux fois par jour). Dans le cas d'une diarrhée sévère persistant malgré un traitement symptomatique, le traitement par Ofev doit être abandonné.
Nausées et vomissements
Les nausées et vomissements sont des effets indésirables fréquemment rapportés (voir «Effets indésirables») et présentent généralement une intensité légère à modérée.
Dans les études INPULSIS (patients FPI), des nausées ont été rapportées chez 24,5% des patients sous Ofev contre 6,6% des patients sous placebo. Les nausées ont entraîné une réduction de la dose d'Ofev chez 1,7% des patients contre aucun des patients sous placebo et l'arrêt du traitement par Ofev chez 2,0% des patients contre aucun des patients sous placebo.
Dans l'étude INBUILD (patients PID fibrosantes progressives), des nausées ont été rapportées chez 28,9% des patients sous Ofev contre 9,4% des patients sous placebo. Les nausées ont entraîné une réduction de la dose d'Ofev chez 3,3% des patients contre 0,6% des patients sous placebo et l'arrêt du traitement par Ofev chez 0,3% des patients contre 0,3% des patients sous placebo.
Dans l'étude SENSCIS (patients PID-ScS), des nausées ont été rapportées chez 31,6% des patients sous Ofev contre 13,5% des patients sous placebo. Les nausées ont entraîné une réduction de la dose d'Ofev chez 2,1% des patients contre aucun des patients sous placebo et l'arrêt du traitement par Ofev chez 2,1% des patients contre aucun des patients sous placebo.
Dans les études INPULSIS (patients FPI), des vomissements ont été rapportés chez 11,6% des patients sous Ofev contre 2,6% des patients sous placebo. Les vomissements ont entraîné une réduction de la dose d'Ofev chez 1,1% des patients contre aucun des patients sous placebo et l'arrêt du traitement par Ofev chez 0,8% des patients contre aucun des patients sous placebo.
Dans l'étude INBUILD (patients PID fibrosantes progressives), des vomissements ont été rapportés chez 18,4% des patients sous Ofev contre 5,1% des patients sous placebo. Les vomissements ont entraîné une réduction de la dose d'Ofev chez 2,4% des patients contre 0,9% des patients sous placebo et l'arrêt du traitement par Ofev chez 0,9% des patients contre aucun des patients sous placebo.
Dans l'étude SENSCIS (patients PID-ScS), des vomissements ont été rapportés chez 24,7% des patients sous Ofev contre 10,4% des patients sous placebo. Les vomissements ont entraîné une réduction de la dose d'Ofev chez 2,1% des patients contre aucun des patients sous placebo et l'arrêt du traitement par Ofev chez 1,4% des patients contre 0,3% des patients sous placebo.
Les vomissements peuvent entraîner une déshydratation avec ou sans troubles électrolytiques, ce qui peut conduire à un dysfonctionnement rénal.
Si les symptômes persistent malgré des mesures de soutien adaptées (incluant un traitement antiémétique), une réduction de la dose ou une interruption du traitement peut devenir nécessaire. Le traitement peut être repris à dose réduite (100 mg deux fois par jour) ou entière (150 mg deux fois par jour). Une persistance de symptômes sévères exige un arrêt du traitement par Ofev.
Fonction hépatique
La sécurité et l'efficacité d'Ofev n'ont pas été étudiées chez les patients présentant une insuffisance hépatique modérée (Child-Pugh B) ou sévère (Child-Pugh C). L'utilisation d'Ofev n'est donc pas recommandée chez ces patients.
Compte tenu de l'augmentation de l'exposition systémique, le risque de survenue d'événements indésirables peut être augmenté chez les patients atteints d'insuffisance hépatique légère (Child Pugh A). Les patients présentant une insuffisance hépatique légère (Child Pugh A) doivent être traités avec une dose d'Ofev réduite (voir «Posologie / Mode d'emploi» et «Pharmacocinétique»).
Des cas de dommages du foie liés à la prise de médicaments ont été observés dans le cadre du traitement par nintédanib. Lors de l'observation du marché, des cas non sévères et sévères de dommages du foie liés à la prise de médicaments ont été signalés, y compris des dommages du foie sévères à issue fatale.
La plupart des événements hépatiques indésirables sont survenus au cours des trois premiers mois de traitement. Par conséquent, les transaminases hépatiques et les concentrations de bilirubine doivent être contrôlées au début du traitement par Ofev, à intervalles réguliers au cours des trois premiers mois du traitement puis périodiquement ou lorsque cliniquement indiqué.
Les augmentations des valeurs d'enzymes hépatiques (ALAT, ASAT, PAL, gamma glutamyl-transférase (GGT)) et de la valeur de bilirubine étaient réversibles dans la plupart des cas après réduction de la dose ou interruption du traitement. Des valeurs accrues d'ALAT et/ou ASAT qui ont augmenté à au moins plus de 3 fois la limite supérieure normale (LSN) ont été observées chez jusqu'à respectivement 5,0%, 7,8% et 4,9% des patients sous Ofev dans les études INPULSIS (patients FPI), INBUILD (patients PID fibrosantes progressives) et SENSCIS (patients PID-ScS), tandis que chez les patients sous placebo, cela concernait moins de 2% des patients au total.
En présence de taux accrus de transaminases (ASAT ou ALAT) à plus de 3 fois la limite supérieure de la normale (LSN), on interrompra l'administration d'Ofev ou réduira la dose à 100 mg deux fois par jour. En même temps, le patient sera étroitement surveillé et les autres causes possibles d'une augmentation des taux d'enzymes hépatiques seront explorées. Après normalisation des taux de transaminases, le traitement par Ofev peut être repris à dose réduite (100 mg deux fois par jour), puis poursuivi à la dose entière (150 mg deux fois par jour). Si l'augmentation des valeurs hépatiques est accompagnée de signes cliniques ou symptômes d'une atteinte hépatique tels qu'un ictère, ou si les taux de transaminases (ASAT ou ALAT) augmentent à plus de 5 fois la LSN, le traitement par Ofev doit être définitivement abandonné (voir «Posologie/Mode d'emploi»).
Les patients à faible poids corporel (< 65 kg), les Asiatiques et les femmes présentent un risque plus élevé d'augmentations des taux d'enzymes hépatiques.
L'exposition au nintédanib a augmenté de façon linéaire avec l'âge des patients. Ceci pourrait également entraîner une hausse du risque d'élévations des enzymes hépatiques (voir «Pharmacocinétique»).
Chez les patients présentant ces facteurs de risque, une surveillance étroite est recommandée.
Fonction rénale
Des cas d'atteinte / insuffisance rénale, dont certains conduisant à la mort, ont été rapportés avec l'utilisation de nintédanib (voir «Effets indésirables»).
Les patients doivent être surveillés pendant le traitement par nintédanib, avec une attention particulière en cas d'existence de facteurs de risque d'atteinte/insuffisance rénale. En cas d'atteinte/insuffisance rénale, une adaptation du traitement doit être envisagée (voir «Ajustement de la posologie»).
Hémorragies
En raison de son mécanisme d'action (inhibition du récepteur du facteur de croissance de l'endothélium vasculaire (vascular endothelial growth factor receptor) VEGFR)), Ofev peut être associé à un risque hémorragique accru.
Dans les études cliniques sur le médicament Ofev, la fréquence des événements hémorragiques chez les patients sous Ofev était légèrement plus élevée ou comparable entre les groupes de traitement (10,3% versus 7,8% dans le groupe placebo dans les études INPULSIS (patients FPI); 11,1% versus 12,7% dans le groupe placebo dans l'étude INBUILD (patients PID fibrosantes progressives), 11,1% versus 8,3% dans le groupe placebo dans l'étude SENSCIS (patients PID-ScS)).
Les saignements les plus fréquemment rapportés étaient une épistaxis sans gravité.
La fréquence des événements hémorragiques graves était faible dans les 2 groupes de traitement (Ofev 1,3% contre placebo 1,4% dans INPULSIS (patients FPI); Ofev 0,9% contre placebo 1,5% dans INBUILD (patients PID fibrosantes progressives); Ofev 1,4% contre placebo 0,7% dans SENSCIS (patients PID-ScS)).
Les études cliniques n'ont pas inclus de patients présentant un risque hémorragique connu (par exemple coagulopathie congénitale ou traitement anticoagulant à dose entière). Des saignements ont été rapportés après la commercialisation, dont certains sévères et mortels (y compris chez des patients avec ou sans traitement par anticoagulants ou autres médicaments pouvant provoquer des saignements). Les patients présentant des risques supplémentaires ne doivent donc être traités par Ofev que si le bénéfice attendu prédomine par rapport au risque potentiel.
Événements thromboemboliques artériels
Les patients ayant récemment subi un infarctus du myocarde ou un accident vasculaire cérébral étaient exclus d'une participation aux études cliniques.
Dans les études cliniques, les événements thromboemboliques artériels ont été rapportés à une faible fréquence (Ofev 2,5% contre placebo 0,7% dans les études INPULSIS (patients FPI); Ofev 0,9% contre placebo 0,9% dans INBUILD (patients PID fibrosantes progressives); Ofev 0,7% contre placebo 0,7% dans SENSCIS (patients PID-ScS)). Dans les études INPULSIS, des infarctus du myocarde ont été décrits chez un pourcentage plus élevé de patients dans le groupe Ofev (1,6%) que dans le groupe placebo, alors que les événements indésirables évoquant une cardiopathie ischémique étaient du même ordre dans le groupe Ofev et dans le groupe placebo. Dans l'étude INBUILD et l'étude SENSCIS, des infarctus du myocarde ont été observés à une faible fréquence: Ofev 0,9% contre placebo 0,9% dans INBUILD; Ofev 0% contre placebo 0,7% dans SCENSIS.
La prudence est de rigueur chez les patients à risque cardiovasculaire accru (par exemple patients présentant une cardiopathie coronarienne connue). Une interruption du traitement doit être envisagée chez les patients développant des signes ou symptômes d'ischémie aiguë du myocarde.
Anévrismes et dissections artérielles
L'utilisation d'inhibiteurs des voies du VEGF chez les patients souffrant ou non d'hypertension peut favoriser la formation d'anévrismes et/ou de dissections artérielles. Avant l'instauration d'Ofev, ce risque doit être soigneusement pris en considération chez les patients présentant des facteurs de risque tels que l'hypertension ou des antécédents d'anévrisme.
Thromboembolies veineuses
Aucune augmentation du risque de thromboembolies veineuses n'a été observée sous nintédanib dans les études cliniques. Le mécanisme d'action du nintédanib pourrait être associé à un risque accru d'événements thromboemboliques.
Perforations gastro-intestinales
Les patients sous nintédanib peuvent avoir un risque accru de perforation gastro-intestinale en raison du mécanisme d'action du médicament. Dans les études INPULSIS (patients FPI), une perforation gastro-intestinale a été rapportée chez 0,3 % des patients sous Ofev et chez aucun (0) des patients sous placebo. Dans l'étude SENSCIS (patients PID-ScS) et dans l'étude INBUILD (patients PID fibrosantes progressives), aucun cas de perforation gastro-intestinale n'a été rapporté dans les deux groupes de traitement.
Après l'introduction sur le marché, des cas de perforations gastro-intestinales, dont certains d'issue fatale, ont été signalés. Une prudence particulière est recommandée chez les patients récemment soumis à une intervention chirurgicale abdominale, en cas de perforation récente d'un organe creux, d'ulcères peptiques dans les antécédents, de diverticulose ou en cas d'utilisation concomitante de corticostéroïdes ou d'AINS. Une perforation gastro-intestinale exige l'arrêt définitif du traitement par Ofev.
Les patients à risque connu de perforation gastro-intestinale ne doivent être traités par Ofev que si le bénéfice attendu prédomine par rapport au risque potentiel.
Troubles de la cicatrisation des plaies
Aucune augmentation de l'incidence des troubles de la cicatrisation n'a été observée dans les études cliniques. Le mécanisme d'action du nintédanib peut perturber la cicatrisation des plaies. Aucune étude n'a été effectuée spécifiquement sur l'impact du nintédanib sur la cicatrisation des plaies. Le traitement par Ofev ne doit donc pas être commencé – ou repris après une interruption due à une intervention chirurgicale – avant que la cicatrisation des plaies soit jugée cliniquement satisfaisante.
Lécithine du soja
Les capsules molles Ofev contiennent de la lécithine du soja (voir «Contre-indications»).
Interactions
Les études effectuées pour identifier les interactions n'ont inclus que des adultes.
Influence d'autres agents actifs sur le nintédanib
Glycoprotéine P (P-gp) et inhibiteurs/inducteurs du CYP3A4
Le nintédanib est un substrat de la P-gp (voir «Pharmacocinétique») et en plus faible mesure un substrat du CYP3A4. Une co-administration de kétoconazole – inhibiteur puissant de la P-gp et inhibiteur du CYP3A4 – a fait augmenter l'AUC du nintédanib d'un facteur 1,61 et la Cmax d'un facteur 1,83.
Chez un patient traité par Ofev, la co-administration d'un inhibiteur puissant de la P-gp (p.ex. kétoconazole ou érythromycine) peut faire augmenter l'exposition au nintédanib. Une surveillance étroite de la tolérance du nintédanib est alors recommandée. Les effets indésirables peuvent exiger une interruption du traitement par Ofev, une réduction de la dose ou un arrêt d'administration d'Ofev (voir «Posologie/Mode d'emploi»).
Une co-administration de rifampicine (inducteur puissant de la P-gp et inducteur du CYP3A4) a fait diminuer l'AUC du nintédanib de 50,3 % et sa Cmax de 60,3 % par rapport à l'administration de nintédanib seul.
Les inducteurs puissants de la P-gp (p.ex. rifampicine, carbamazépine, phénytoïne et millepertuis) peuvent faire diminuer l'exposition au nintédanib. Le choix d'une co-médication alternative dont le potentiel d'induction de la P-gp est faible ou inexistant doit être envisagé.
Aliments
Ofev doit être pris avec un repas pour améliorer la biodisponibilité (voir «Pharmacocinétique»).
pH
La solubilité du nintédanib dépend fortement du pH et baisse lors d'un pH >3. Dans les études cliniques, la co-administration de médicaments causant une augmentation du pH intragastrique n'a pas influencé les valeurs de Ctrough du nintédanib pris avec un repas. Lors d'une prise à jeun, la co-administration de médicaments induisant une augmentation du pH intragastrique pourrait réduire la biodisponibilité du nintédanib. Le nintédanib doit donc être pris avec des aliments.
Tabagisme
Le tabagisme est associé à une diminution de l'exposition au nintédanib, susceptible d'altérer son efficacité. Les patients doivent être incités à arrêter de fumer avant de commencer le traitement par Ofev ou à réduire leur consommation de tabac pendant le traitement par Ofev.
D'après les études in vitro, le nintédanib n'est pas un substrat des transporteurs OATP-1B1, OATP-1B3, OATP-2B1, OCT-2, BCRP ou MRP-2. Des études in vitro ont révélé que le nintédanib est un substrat du transporteur OCT-1. On estime que ces observations n'ont guère de signification clinique.
Influence du nintédanib sur d'autres agents actifs
Transporteurs
De faibles effets inhibiteurs sur les transporteurs OCT-1, BCRP et P-gp ont été observés in vitro. La signification clinique de ces effets n'a pas été étudiée, mais elle est considérée comme faible.
D'après les études in vitro, le nintédanib n'est pas un inhibiteur des transporteurs OATP-1B1, OATP-1B3, OATP-2B1, OCT-2 ou MRP-2.
Il est apparu que le métabolite BIBF 1202 était in vitro un faible inhibiteur des transporteurs OATP-1B1, OATP-1B3, OATP-2B1, OCT-1 et BCRP. La signification clinique de ces effets n'a pas été étudiée, mais elle est considérée comme faible.
Enzymes du cytochrome (CYP)
La biodégradation du nintédanib ne s'effectue qu'en faible mesure par voie des enzymes du CYP. Le nintédanib et ses métabolites – le groupement acide libre BIBF 1202 et le BIBF 1202 glucuronidé – n'ont pas eu d'effet inhibiteur ou inducteur sur les enzymes du CYP dans les études précliniques (voir «Pharmacocinétique»). On estime donc que la probabilité d'interactions médicamenteuses dues à une influence du nintédanib sur la voie métabolique du CYP est faible.
Administration en association avec d'autres médicaments
Traitement concomitant par pirfénidone
Les données issues d'une étude spécifique de pharmacocinétique n'ont pas mis en évidence d'interaction pharmacocinétique significative entre le nintédanib et la pirfénidone lorsque ces principes actifs sont administrés en association.
Traitement concomitant par bosentan
Dans une étude spéciale de pharmacocinétique, le traitement concomitant par Ofev associé à du bosentan a été étudié sur des sujets volontaires sains. Les participants à l'étude ont reçu une dose unique de 150 mg d'Ofev avant et après une administration répétée de 125 mg de bosentan deux fois par jour à l'état d'équilibre. Les rapports ajustés des valeurs moyennes géométriques (intervalle de confiance (IC) à 90 %) étaient respectivement de 103 % (86 % - 124 %) et 99 % (91 % - 107 %) pour la Cmax et l'AUC0-tz du nintédanib (n=13), ce qui indique que l'administration concomitante du nintédanib avec du bosentan n'a pas modifié la pharmacocinétique du nintédanib.
Le potentiel d'interactions entre le nintédanib et les contraceptifs hormonaux n'a pas été étudié.
Grossesse/Allaitement
Grossesse
On ne dispose pas de données sur l'utilisation d'Ofev chez la femme enceinte, mais les expérimentations animales précliniques ont révélé une toxicité du médicament sur la reproduction (voir «Données précliniques»). Vu que le nintédanib peut nuire également au fœtus humain, il ne doit pas être utilisé pendant la grossesse.
Un test de grossesse doit être effectué avant le début du traitement par Ofev et être répété si nécessaire pendant le traitement.
Il faut informer les patientes qu'elles devront prévenir leur médecin ou leur pharmacien si elles constatent le début d'une grossesse au cours de leur traitement par Ofev.
Si une patiente est enceinte pendant le traitement par Ofev, le traitement doit être arrêté et la patiente doit être informée des risques potentiels pour le fœtus.
Contraception
Le nintédanib peut nuire au fœtus (voir la rubrique «Données précliniques»).
Les patientes susceptibles de procréer qui sont traitées par Ofev doivent être averties de la nécessité d'éviter une grossesse pendant le traitement par Ofev et que, pendant et jusqu'à 3 mois après la dernière dose d'Ofev, des méthodes contraceptives très efficaces doivent être utilisées.
Étant donné qu'actuellement, on ne sait pas si le nintédanib peut diminuer l'efficacité des moyens de contraception hormonaux, les femmes doivent utiliser une méthode barrière en plus des contraceptifs hormonaux.
Allaitement
On ne dispose pas de données concernant le passage du nintédanib et de ses métabolites dans le lait maternel.
L'excrétion de faibles quantités de nintédanib et de ses métabolites (≤0,5 % de la dose administrée) dans le lait des rates allaitantes a été constatée dans des études précliniques.
Un risque pour le nouveau-né / l'enfant ne peut pas être exclu. L'allaitement doit être interrompu pendant le traitement par Ofev.
Effet sur l’aptitude à la conduite et l’utilisation de machines
Aucune étude correspondante n'a été effectuée.
Les patients doivent être instruits d'être prudents en conduisant un véhicule ou en utilisant une machine au cours du traitement par Ofev.
Effets indésirables
La sécurité d'Ofev a été évaluée dans des études cliniques auprès de plus de 1000 patients atteints de FPI, dont plus de 200 ont reçu Ofev plus de 2 ans. Chez des patients atteints de FPI, Ofev a été évalué dans trois études de 52 semaines avec randomisation, double aveugle et contrôle versus placebo. Dans l'étude de phase II (TOMORROW) et les études de phase III (INPULSIS-1 et INPULSIS-2), 723 patients atteints de FPI ont reçu Ofev à la dose de 150 mg deux fois par jour et 508 patients ont reçu un placebo. La durée médiane d'exposition a été de 10 mois chez les patients traités par Ofev et de 11 mois chez les patients sous placebo. Les participants aux études étaient âgés de 42 à 89 ans (âge médian: 67 ans). La majorité des patients étaient de sexe masculin (79 %) et de type caucasoïde (60 %).
Ofev a été en outre évalué dans une étude de phase III, randomisée, menée en double aveugle et contrôlée contre placebo (INBUILD), dans laquelle le traitement par Ofev 150 mg deux fois par jour (n=332) a été comparé avec le placebo chez 663 patients atteints d'autres PID fibrosantes chroniques avec un phénotype progressif. Alors que les patients ont été traités pendant au moins 52 semaines, le traitement a duré jusqu'à 27 mois chez certains patients. La durée médiane d'exposition était de 16 mois chez les patients sous Ofev et de 17 mois chez les patients sous placebo. L'âge des participants à l'étude allait de 27 à 87 ans (âge médian: 67 ans). La plupart des patients étaient de sexe masculin (54%). Les patients étaient majoritairement caucasiens (74%) ou asiatiques (25%).
Ofev a été évalué dans une étude de phase III avec randomisation, double aveugle et contrôle versus placebo (SENSCIS) dans laquelle 576 patients atteints de PID-ScS ont reçu deux fois par jour 150 mg d'Ofev (n=288) ou le placebo (n=288). Tandis que les patients ont été traités durant au moins 52 semaines, le traitement de certains patients a duré jusqu'à 100 semaines. La durée d'exposition médiane était de 15 mois pour les patients sous Ofev et 16 mois pour les patients sous placebo. L'âge des participants allait de 20 à 79 ans (âge médian: 55 ans). La plupart des patients étaient de sexe féminin (75 %). Les patients étaient majoritairement caucasiens (67 %), asiatiques (25 %) ou avaient la peau noire (6 %). 49 % des patients ont reçu un traitement stable par mycophénolate au début de l'étude.
Les effets indésirables le plus souvent décrits en rapport avec l'administration d'Ofev étaient des diarrhées, nausées et vomissements, des douleurs abdominales, une perte d'appétit, une perte de poids, des hémorragies, des taux accrus d'enzymes hépatiques et une éruption cutanée.
Le profil de sécurité était similaire chez les patients traités sous Ofev, et ce indépendamment du fait qu'ils aient reçu du mycophénolate au début de l'étude ou non.
La section «Mises en garde et précautions» décrit des effets indésirables sélectionnés et la marche à suivre correspondante.
Les catégories de fréquence des effets indésirables sont définies selon la convention suivante: «très fréquents» (≥1/10), «fréquents» (< 1/10, ≥1/100), «occasionnels» (< 1/100, ≥1/1000), «rares» (< 1/1000, ≥1/10'000), «très rares» (< 1/10.000).
Fibrose pulmonaire idiopathique (FPI)
Affections hématologiques et du système lymphatique
Occasionnels: thrombocytopénie.
Troubles du métabolisme et de la nutrition
Fréquents: perte d'appétit, perte de poids.
Affections du système nerveux
Fréquents: céphalées.
Affections vasculaires
Fréquents: hémorragiesa.
Occasionnels: hypertensionb.
Fréquence non connue: anévrismes et dissections artérielles.
Affections gastro-intestinales
Très fréquents: diarrhées (53,6%), nausées (19,1%), douleurs abdominalesc (10,2%).
Fréquents: vomissements.
Occasionnels: pancréatites, perforations gastro-intestinales, colites.
Affections hépatobiliaires
Très fréquents: augmentation des enzymes hépatiques (10,5%).
Fréquents: augmentation de l'alanine aminotransférase (ALAT), augmentation de l'aspartate aminotransférase (ASAT), augmentation de la gamma-glutamyltransférase (GGT).
Occasionnels: augmentation du taux sanguin des phosphatases alcalines (PAL), hyperbilirubinémie, atteinte hépatique d'origine médicamenteuse.
Affections de la peau et du tissu sous-cutané
Fréquents: éruption cutanée.
Occasionnels: prurit, alopécie.
Affections du rein et des voies urinaires
Fréquence non connue: insuffisance rénale (voir «Mises en garde et précautions»).
a Des évènements hémorragiques sévères et non sévères, dont certains mortels, correspondant à l'expérience acquise pendant les études cliniques, ont été observés après la mise sur le marché. Les évènements hémorragiques les plus fréquents comprenaient des saignements rectaux/hématochézie, des saignements de nez et des hématomes. Les événements mortels concernaient des saignements gastro-intestinaux, intracrâniens et pulmonaires ainsi que des CIVD.
b Incluant: hypertension, tension artérielle accrue, poussée hypertensive et cardiopathie hypertensive.
c Incluant: douleurs abdominales, douleurs épigastriques, douleurs pelviennes, douleurs gastro-intestinales, sensation douloureuse de pression abdominale.
Autres pneumopathies interstitielles diffuses (PID) fibrosantes chroniques avec un phénotype progressif
Affections hématologiques et du système lymphatique
Occasionnels: thrombocytopénie.
Troubles du métabolisme et de la nutrition
Très fréquents: perte d'appétit (11,1%).
Fréquents: perte de poids.
Affections du système nerveux
Fréquents: céphalées.
Affections vasculaires
Fréquents: hypertension, hémorragies.
Fréquence non connue: anévrismes et dissections artérielles.
Affections gastro-intestinales
Très fréquents: diarrhées (59%), nausées (24%), douleurs abdominales (11%), vomissements (12%).
Occasionnels: pancréatites, colites.
Fréquence non connue: perforation gastro-intestinale.
Affections hépatobiliaires
Très fréquents: augmentation des enzymes hépatiques (18,4%), augmentation de l'alanine aminotransférase (ALAT) (10,8%).
Fréquents: augmentation de l'aspartate aminotransférase (ASAT), augmentation de la gamma-glutamyl transférase (GGT), augmentation du taux sanguin des phosphatases alcalines (PAL), atteinte hépatique d'origine médicamenteuse.
Occasionnels: hyperbilirubinémie
Affections de la peau et du tissu sous-cutané
Fréquents: éruption cutanée.
Occasionnels: prurit, alopécie.
Affections du rein et des voies urinaires
Fréquence non connue: insuffisance rénale (voir «Mises en garde et précautions»).
Pneumopathie interstitielle associée à la sclérodermie systémique (PID-ScS)
Affections hématologiques et du système lymphatique
Occasionnels: thrombocytopénie.
Troubles du métabolisme et de la nutrition
Fréquents: perte d'appétit, perte de poids.
Affections du système nerveux
Fréquents: céphalées.
Affections vasculaires
Fréquents: hypertension, hémorragies.
Fréquence non connue: anévrismes et dissections artérielles.
Affections gastro-intestinales
Très fréquents: diarrhées (68,4%), nausées (24,7%), douleurs abdominales (11,8%), vomissements (17,7%).
Occasionnels: colites.
Fréquence non connue: pancréatites, perforations gastro-intestinales.
Affections hépatobiliaires
Très fréquents: augmentation des enzymes hépatiques (11,1%).
Fréquents: augmentation de l'alanine aminotransférase (ALAT), augmentation de l'aspartate aminotransférase (ASAT), augmentation de la gamma-glutamyltransférase (GGT).
Occasionnels: atteinte hépatique d'origine médicamenteuse.
Fréquence non connue: hyperbilirubinémie.
Affections de la peau et du tissu sous-cutané
Occasionnels: éruption cutanée, prurit.
Fréquence non connue: alopécie.
Affections du rein et des voies urinaires
Occasionnels: insuffisance rénale (voir «Mises en garde et précautions»).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Il n'existe aucun antidote spécifique et aucun traitement spécifique d'un surdosage d'Ofev. La plus forte dose unique de nintédanib administrée dans une étude de phase I était de 450 mg une fois par jour. À part cela, 2 patients ont reçu une surdose ne dépassant pas 600 mg deux fois par jour pendant une période allant jusqu'à 8 jours. Les effets indésirables observés étaient en accord avec le profil de sécurité connu du nintédanib, comprenant des taux accrus d'enzymes hépatiques et des symptômes gastro-intestinaux. Les deux patients se sont rétablis de ces effets indésirables.
Dans le cadre des études INPULSIS (patients FPI), un patient reçu accidentellement une dose de 600 mg par jour pendant 21 jours au total. Un effet indésirable sans gravité (rhinopharyngite) est apparu et a régressé au cours de la période de surdosage. Aucun autre événement décrit ne s'est produit.
Lors d'un surdosage, le traitement doit être interrompu et des mesures générales de soutien adaptées à la situation clinique doivent être initiées.
Propriétés/Effets
Code ATC
L01XE31
Mécanisme d'action
Le nintédanib est un inhibiteur de tyrosines kinases qui appartient au type des petites molécules. Les tyrosine kinases inhibées comprennent les récepteurs du facteur de croissance dérivé des plaquettes (platelet-derived growth factor receptors, PDGFR) α et β, les récepteurs du facteur de croissance des fibroblastes (fibroblast growth factor receptors, FGFR) 1 à 3 et les récepteurs du facteur de croissance de l'endothélium vasculaire (vascular endothelial growth factor receptors, VEGFR) 1 à 3. Le nintédanib se fixe de manière compétitive au site de liaison de l'adénosine triphosphate (ATP) de ces récepteurs et bloque ainsi la signalisation intracellulaire qui est décisive pour la prolifération, la migration et la transformation des fibroblastes, et donc pour les mécanismes pathologiques de la FPI. À part cela, le nintédanib inhibe aussi les kinases Flt-3, Lck, Lyn et Src.
Pharmacodynamique
L'activation des cascades de signalisation induites par le FGFR et le PDGFR est un facteur décisif pour la prolifération et la migration des fibroblastes/myofibroblastes pulmonaires, qui sont les cellules caractéristiques de la pathologie de fibrose pulmonaire idiopathique. La signification potentielle de l'inhibition du VEGFR pour la pathologie de FPI n'a pas encore été totalement élucidée. On suppose que le nintédanib, en se liant au site de liaison d'adénosine triphosphate (ATP) du domaine intracellulaire des récepteurs à activité tyrosine kinase, inhibe à l'échelle moléculaire les cascades de signalisation du FGFR et du PDGFR impliquées dans la prolifération et la migration des fibroblastes pulmonaires, et interfère ainsi avec l'activation croisée (par autophosphorylation du récepteur homodimère).
In vitro, des concentrations faiblement nanomolaires de nintédanib suffisent pour inhiber les récepteurs cibles. Sur des fibroblastes pulmonaires de patients atteints de FPI, la prolifération stimulée par PDGF, FGF et VEGF a été inhibée par le nintédanib aux CE50 de 11 nmol/l, 5,5 nmol/l et moins de 1 nmol/l. À des concentrations de 100 à 1000 nmol/l, le nintédanib a également inhibé la migration de fibroblastes stimulée par PDGF, FGF et VEGF ainsi que la transformation des fibroblastes en myofibroblastes induite par TGF-β2. On suppose que l'activité anti-inflammatoire du nintédanib limite en outre la stimulation fibrotique en réduisant les médiateurs profibrotiques tels que l'IL-1β et l'IL-6. On ignore encore dans quelle mesure l'activité anti-angiogénique du nintédanib participe au mécanisme d'action du médicament dans le traitement des fibroses pulmonaires. Dans des études in vivo, le nintédanib a fait preuve d'une forte activité anti-fibrotique et anti-inflammatoire.
Efficacité clinique
Fibrose pulmonaire idiopathique (FPI)
L'efficacité clinique du nintédanib a été étudiée chez 1231 patients atteints de FPI dans une étude de phase II (TOMORROW) et deux études de phase III (INPULSIS-1 et INPULSIS-2). Il s'agissait d'études randomisées, en double aveugle, avec contrôle contre placebo, dans lesquelles un traitement de 52 semaines par Ofev (150 mg deux fois par jour) a été évalué versus placebo.
Les études INPULSIS-1 et INPULSIS-2 étaient de conception identique, proche de la conception de l'étude TOMORROW. Les patients ont été randomisés en proportions de 3:2 (1:1 dans l'étude TOMORROW) et assignés pour 52 semaines à un traitement par Ofev (150 mg deux fois par jour) ou par un placebo (également deux fois par jour). L'étude TOMORROW comprenait des groupes de traitement supplémentaires (50 mg par jour, 50 mg deux fois par jour et 100 mg deux fois par jour) qui ne doivent pas être discutés ici.
Le critère primaire était défini comme le déclin annuel de la capacité vitale forcée (CVF). La variation du score total au questionnaire SGRQ (Saint George's Respiratory Questionnaire) entre le début de l'étude et la semaine 52 ainsi que le temps écoulé jusqu'à la première exacerbation aiguë de la FPI étaient des critères secondaires importants des études INPULSIS et des critères secondaires de l'étude TOMORROW.
Déclin annuel de la CVF
Le déclin annuel de la CVF (en ml) était significativement plus faible chez les patients sous nintédanib que chez les patients sous placebo. L'effet du traitement était concordant entre les 3 études. Le tableau 1 présente les résultats des études individuelles et les résultats de l'analyse cumulée des études INPULSIS.
Tableau 1: Déclin annuel de la CVF (ml) dans les études TOMORROW, INPULSIS-1 et INPULSIS-2 ainsi que dans l'analyse cumulée des études INPULSIS – population traitée2
TOMORROW | INPULSIS-1 | INPULSIS-2 | INPULSIS-1 et INPULSIS-2 Données réunies | |||||
|---|---|---|---|---|---|---|---|---|
Placebo | Ofev 150 mg deux fois par jour | Placebo | Ofev 150 mg deux fois par jour | Placebo | Ofev 150 mg deux fois par jour | Placebo | Ofev 150 mg deux fois par jour | |
Nombre de patients inclus à l'analyse | 83 | 84 | 204 | 309 | 219 | 329 | 423 | 638 |
Taux 1,2 (ET) de déclin sur 52 semaines | -190 (36) | -60 (39) | −239,9 (18,71) | −114,7 (15,33) | −207,3 (19,31) | −113,6 (15,73) | −223,5 (13,45) | −113,6(10,98) |
Comparaison vs placebo | ||||||||
Différence1 | 131 | 125,3 | 93,7 | 109,9 | ||||
IC à 95 % | (27; 235) | (77,7; 172,8) | (44,8; 142,7) | (75,9; 144,0) | ||||
Valeur p | 0,01363 | <0,0001 | 0,0002 | <0,0001 | ||||
1 Calcul sur la base d'un modèle de régression à coefficient aléatoire. 2 Population randomisée pour l'étude TOMORROW; population traitée pour l'étude INPULSIS-1, l'étude INPULSIS-2 et les données réunies des études INPULSIS 3 Valeur p nominale. | ||||||||
Toutes les analyses de sensibilité prédéfinies ont confirmé la robustesse de l'effet du nintédanib en termes de réduction du taux annuel de déclin de la CVF. À part cela, des effets comparables sur d'autres critères de la fonction pulmonaire – par exemple la variation de la CVF à 52 semaines versus valeur initiale et l'analyse des répondeurs en termes de CVF – ont été observés, confirmant les effets ralentissants du nintédanib sur la progression de la maladie. La figure 1 présente l'évolution de la CVF en fonction du temps par rapport aux valeurs initiales pour les deux groupes de traitement (sur la base de l'analyse cumulée des études INPULSIS-1 et INPULSIS-2).
Figure 1: Variation moyenne observée (ETM) de la CVF versus valeur initiale (ml) en fonction du temps – études INPULSIS-1 et INPULSIS-2 (données réunies)
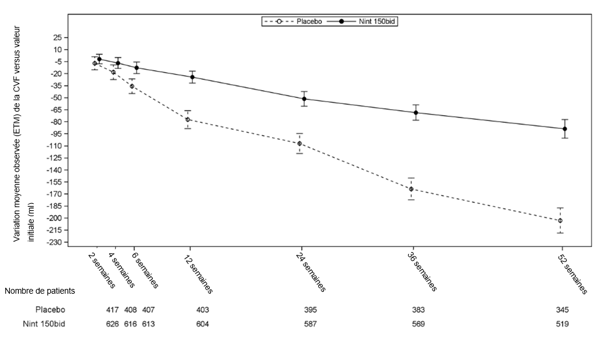
bid = deux fois par jour, ETM = erreur type de la moyenne
Variation du score SGRQ total à 52 semaines versus valeur initiale
Le score total au SGRQ (St. George's Respiratory Questionnaire) est une mesure de la qualité de vie en fonction de la santé (HRQoL). Il a été analysé à 52 semaines. Dans l'étude TOMORROW, la variation moyenne estimée du score SGRQ total entre le début de l'étude (valeur initiale) et la semaine 52 était de 5,46 sous placebo, indiquant une détérioration de la qualité de vie liée à la santé, tandis qu'elle était de -0,66 sous nintédanib, indiquant une stabilité de la qualité de vie liée à la santé. La différence moyenne estimée entre le nintédanib et le placebo était de -6,12 (IC à 95 %: -10,57; -1,67; p = 0,0071).
Dans l'étude INPULSIS-2, l'augmentation du score SGRQ total par rapport à la valeur initiale était plus prononcée chez les patients sous placebo que chez ceux traités par le nintédanib 150 mg deux fois par jour. La détérioration de la HRQoL était plus légère dans le groupe sous nintédanib. La différence entre les deux groupes de traitement était statistiquement significative (-2,69; IC à 95 %: -4,95; -0,43; p = 0,0197).
Dans l'étude INPULSIS-1, l'augmentation du score SGRQ total à 52 semaines versus valeur initiale était similaire sous nintédanib et sous placebo (différence entre les groupes de traitement: -0,05; IC à 95 %: -2,50; 2,40; p = 0,9657). Dans l'analyse cumulée des études INPULSIS, la variation moyenne estimée du score SGRQ total à 52 semaines versus valeur initiale était plus faible sous nintédanib (3,53) que sous placebo (4,96). La différence entre les traitements était de -1,43 (IC à 95 %: -3,09; 0,23; p = 0,0923). L'influence du nintédanib sur la qualité de vie liée à la santé, évaluée à l'aide du score SGRQ total, est globalement modérée et correspond à une plus faible détérioration que sous placebo.
Temps écoulé jusqu'à la première exacerbation aiguë de la FPI
Dans les études TOMORROW et INPULSIS-2, le risque de subir une première exacerbation aiguë de la FPI au cours de la période d'observation de 52 semaines était significativement plus faible sous nintédanib que sous placebo (hazard ratio [HR]: 0,16; IC à 95 %: 0,04; 0,71; p = 0,0054) et (HR: 0,38; IC 95 %: 0,19; 0,77; p = 0,005). Dans l'étude INPULSIS-1, aucune différence n'a été constatée entre les groupes de traitement (HR: 1,15; IC à 95 %: 0,54; 2,42; p = 0,6728).
Analyse de survie
Dans l'analyse cumulée prédéfinie des données de survie issues des études INPULSIS, le taux de mortalité globale au cours des 52 semaines était numériquement plus faible sous nintédanib (5,5 %) que sous placebo (7,8 %). L'analyse du temps écoulé jusqu'au décès a révélé un HR de 0,70 (IC à 95 % 0,43; 1,12; p = 0,1399). Les résultats de tous les critères d'évaluation concernant la survie (tels que la mortalité pendant le traitement et la mortalité de cause respiratoire) ont montré systématiquement une différence numérique en faveur du nintédanib, mais sans atteindre le seuil de signification statistique.
Données complémentaires issues de l'étude de phase IV INJOURNEY associant Ofev 150 mg deux fois par jour et la pirfénidone:
Au cours d'un essai exploratoire, en ouvert, randomisé, l'administration concomitante de 150 mg de nintédanib deux fois par jour et de pirfénidone (augmentation progressive de la dose jusqu'à 801 mg trois fois par jour) a été comparée à l'administration de nintédanib 150 mg deux fois par jour seul chez un total de 105 patients pendant 12 semaines. Le critère d'évaluation principal était le pourcentage de patients présentant des évènements indésirables gastro-intestinaux entre le début de l'essai et la semaine 12.
Les évènements gastro-intestinaux étaient fréquents et correspondaient au profil de sécurité connu de chaque composant. Les diarrhées, les nausées et les vomissements étaient les évènements indésirables les plus fréquemment rapportés, soit respectivement 20 patients (37,7 %) contre 16 patients (31,4 %), 22 patients (41,5 %) contre 6 patients (11,8 %) et 15 patients (28,3 %) contre 6 patients (11,8 %) traités par l'ajout de pirfénidone au nintédanib comparativement au traitement par nintédanib seul.
Les variations moyennes absolues (ET) de la CVF à la semaine 12 par rapport aux valeurs initiales étaient de −13,3 (17,4) ml chez les patients traités par nintédanib avec ajout de pirfénidone (n = 48), comparé à −40,9 (31,4) ml chez les patients traités par nintédanib seul (n = 44).
Pneumopathies interstitielles diffuses (PID) fibrosantes chroniques avec un phénotype progressif
L'efficacité clinique d'Ofev a été évaluée dans une étude de phase III randomisée, menée en double aveugle et contrôlée contre placebo (INBUILD) chez des patients atteints de PID fibrosante chronique avec un phénotype progressif.
Au total, 663 patients ont été randomisés selon un rapport 1:1 pour recevoir soit 2x150 mg d'Ofev par jour, soit le placebo pendant au moins 52 semaines.
La randomisation était stratifiée sur l'aspect de la fibrose à la tomodensitométrie à haute résolution (TDM-HR). 412 patients avec un aspect de fibrose de type pneumopathie interstitielle commune (PIC) et 251 patients avec d'autres aspects de fibrose ont été randomisés. Deux populations co-primaires ont été définies pour l'évaluation de cette étude: la population globale et la population avec un aspect de fibrose de type PIC à la TDM-HR.
Le critère d'évaluation principal était le taux annuel de déclin de la CVF (en ml) sur 52 semaines. D'autres critères d'évaluation étaient, par exemple, le délai de survenue de la première exacerbation aiguë de la PID ou du décès ou le délai de survenue du décès.
Les patients ayant un diagnostic clinique de PID fibrosante chronique étaient éligibles à l'étude s'ils présentaient une fibrose significative (>10% de lésions fibrotiques) à la TDM-HR ainsi que des signes cliniques de progression (définie comme un déclin de la CVF ≥10%, un déclin de la CVF entre 5% et 10% avec aggravation des symptômes OU aggravation à l'imagerie, ou une aggravation des symptômes ET une aggravation à l'imagerie) au cours des 24 mois précédents malgré un traitement adéquat. Les patients devaient présenter une CVF ≥45% de la valeur prédite et une DLCO comprise entre 30% et 80% de la valeur prédite.
Étaient exclus de la participation à l'étude les patients avec
- FPI, bronchoconstriction pertinente (p.ex. VEMS/CVF <0,7 avant bronchodilatation) ou hypertension pulmonaire significative
- élévation des transaminases ou de la bilirubine >1,5 LSN
- risque d'hémorragie connu, anticoagulation complète, infarctus du myocarde récent ou accident vasculaire cérébral récent
- traitement antérieur par nintédanib, pirfénidone, azathioprine, ciclosporine, mycophénolate, tacrolimus, corticoïdes oraux >20 mg/jour ou corticoïdes oraux+azathioprine-N-acétylcystéine dans les 4 semaines avant la randomisation*
- traitement antérieur par cyclophosphamide dans les 8 semaines avant la randomisation*
- traitement antérieur par rituximab dans les 6 mois avant la randomisation*
* L'utilisation des médicaments mentionnés était à nouveau autorisée au bout de 6 mois après le début de l'étude, pour autant que ceci soit cliniquement indiqué.
Les participants à l'étude étaient majoritairement caucasiens (74%) ou asiatiques (25%). 54% des patients étaient de sexe masculin, 49% n'avaient jamais fumé, l'âge moyen était de 66 ans et la CVF s'élevait en moyenne à 69% de la valeur prédite. Les PID sous-jacentes étaient des pneumopathies d'hypersensibilité (26%), des PID auto-immunes (26%), des pneumopathies interstitielles non spécifiques idiopathiques (19%), des pneumopathies interstitielles idiopathiques inclassifiables (17%) et d'autres PID (12%).
Taux annuel de déclin de la CVF (ml)
Une réduction statistiquement significative du taux de déclin de la CVF sur 52 semaines a été observée sous traitement par Ofev contre placebo. La différence constatée entre Ofev et placebo était de 107 ml (Tableau 2).
Tableau 2. Taux annuel de déclin de la CVF (ml, critère d'évaluation principal de l'étude INBUILD)
Population totale | Sous-population de type PIC | Sous-population Autres aspects de fibrose à la TDM-HR | ||||
|---|---|---|---|---|---|---|
Ofev | Placebo | Ofev | Placebo | Ofev | Placebo | |
Nombre de patients évalués | 332 | 331 | 206 | 206 | 126 | 125 |
Tauxa (ES) de déclin sur 52 semaines | -80,8 | -187,8 | -82,9 | -211,1 | -79,0 | -154,2 |
Comparaison contre placebo | 107,0 | 128,2 | 75,2* | |||
IC à 95% | (65,4, 148,5) | (70,8, 185,6) | (15,5, 135,0)* | |||
Valeur p | < 0,0001 | < 0,0001 | ||||
* Une comparaison reposant sur la sous-population avec d'autres aspects de fibrose à la TDM-HR n'a pas été reprise dans la procédure de tests multiples. Les valeurs présentées ici servent à des fins descriptives. a Basé sur un modèle de régression avec coefficients aléatoires et les effets de catégories fixes du traitement, l'aspect TDM-HR, les effets continus fixes du temps, la valeur initiale de la CVF (ml), y compris l'interaction entre le traitement et le temps et l'interaction entre la valeur initiale et le temps. | ||||||
La Figure 2 montre l'évolution de la variation de la CVF dans le temps par rapport à la valeur initiale, dans les groupes de traitement.
Figure 2: Variation moyenne observée (ETM) de la CVF par rapport à la valeur initiale (ml) sur 52 semaines
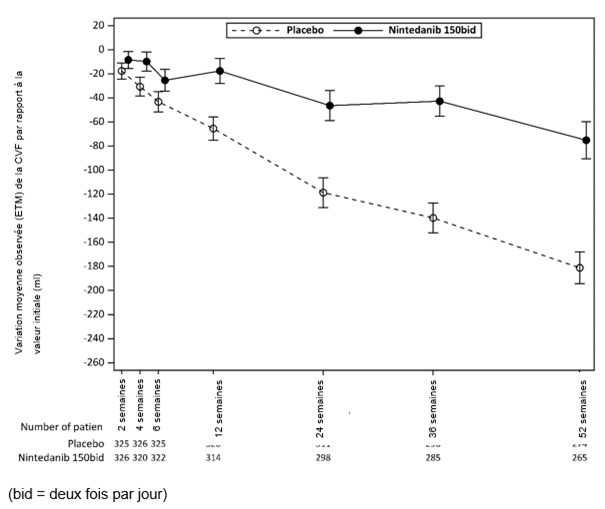
Une analyse exploratoire post-hoc réalisée selon le diagnostic de PID a été effectuée (Figure 3). Dans l'ensemble, un effet thérapeutique cohérent a été mis en évidence au sein des différents diagnostics de PID.
Figure 3: Taux annuel de déclin de la CVF (ml) sur 52 semaines reposant sur le diagnostic de base de PID dans l'étude 5*
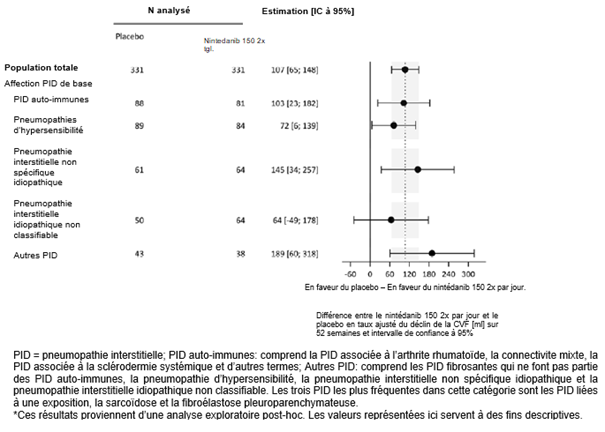
Déclin de la CVF en pourcentage
La Figure 4 représente la variation en pourcentage de la CVF (ml) de la valeur initiale à la semaine 52. La dégradation de la fonction pulmonaire était moins importante chez la majorité des patients sous Ofev que chez les patients sous placebo.
Figure 4: Histogramme de la variation en pourcentage de la CVF (ml) de la valeur initiale à la semaine 52 après traitement avec incréments (%) ou décréments par paliers de 5 (étude 5)a
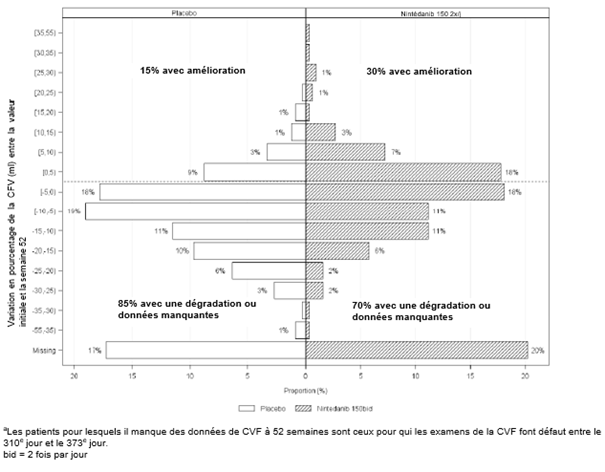
Délai de survenue de la première exacerbation aiguë de la PID ou du décès
Le risque d'une première exacerbation aiguë de la PID ou de décès a diminué dans le groupe Ofev par rapport au groupe placebo sur 52 semaines (HR Ofev contre placebo: 0,80; IC à 95%: 0,48; 1,34) ou sur toute la durée de l'étude (HR Ofev contre placebo: 0,67; IC à 95%: 0,46; 0,98), ce qui correspond à une réduction de 33% du risque d'une première exacerbation aiguë de la PID ou de décès chez les patients sous Ofev par comparaison avec le placebo.
Analyse de la survie
Dans l'étude INBUILD, les données de survie sous Ofev par comparaison avec le placebo ont été évaluées afin d'étayer le critère d'évaluation principal (CFV). La mortalité toutes causes confondues a été évaluée sur toute la durée de l'étude et sur la période de suivi disponible, indépendamment de la cause du décès et de la poursuite ou non du traitement. L'analyse n'a pas montré de différence statistiquement significative sur 52 semaines (HR Ofev contre placebo 0,94, IC à 95% 0,47, 1,86), et sur toute la durée de l'étude (HR Ofev contre placebo 0,78, IC 95% 0,5, 1,21).
Pneumopathie interstitielle associée à la sclérodermie systémique (PID-ScS)
L'efficacité clinique du nintédanib a été évaluée dans une étude de phase III randomisée, en double aveugle et contrôlée versus placebo (SENSCIS) chez des patients atteints de PID-ScS.
En tout, 580 patients avec une proportion de 1:1 ont été randomisés pour recevoir soit 150 mg d'Ofev deux fois par jour ou le placebo pendant au moins 52 semaines. La randomisation a été stratifiée selon le statut des anticorps anti-topoisomérase (ATA). Certains patients ont poursuivi le traitement en aveugle durant jusqu'à 100 semaines.
Le diagnostic des patients atteints de PID-ScS reposait sur les critères de classification pour la sclérodermie systémique (ScS) de l'American College of Rheumatology / de la European League Against Rheumatism lorsque la survenue de la maladie (premier symptôme non Raynaud) remontait à moins de 7 ans et en présence d'une fibrose pulmonaire d'au moins 10%, constatée sur la base d'une image obtenue par tomodensitométrie haute résolution effectuée au cours des 12 mois précédents.
Chez les patients, la CVF devait atteindre au moins 40 % de la valeur estimée, tandis que la DLCO devait être entre 30 et 89 % de la valeur estimée. Les patients atteints d'importantes obstructions des voies respiratoires (c'est-à-dire VEMS/CVF avant la bronchodilatation inférieur à 0,7) ou ayant reçu ou allant recevoir une transplantation de cellules souches hématopoïétiques ne pouvaient pas participer à l'étude. D'autres critères d'exclusion étaient des valeurs ALAT, ASAT ou de bilirubine élevées (>1,5xLNS), un risque accru d'hémorragies (y compris avec anticoagulation thérapeutique), un infarctus du myocarde ou un accident vasculaire cérébral récent, une hypertension pulmonaire considérable, des ulcérations à au moins trois bouts de doigt, un antécédent de nécrose sévère du doigt avec hospitalisation nécessaire ainsi qu'un antécédent de crise rénale sclérodermique. Les patients qui recevaient d'autres médicaments à l'étude, qui avaient déjà été traités par nintédanib ou pirfénidone et avaient reçu de l'azathioprine 8 semaines avant la randomisation ou du cyclophosphamide ou de la cyclosporine A 6 mois avant la randomisation ont également été exclus. Un traitement stable par mycophénolate ou méthotrexate ainsi que par prednisone ≤10 mg/jour ou une dose équivalente était autorisé.
Le critère d'évaluation primaire était le déclin annuel de la capacité vitale forcée (CVF) sur 52 semaines.
Les critères secondaires importants étaient la variation absolue du Rodnan Skin Score modifié (mRSS) à la semaine 52 par rapport à la valeur initiale, la variation absolue du score total au Saint George's Respiratory Questionnaire (SGRQ) à la semaine 52 par rapport à la valeur initiale ainsi que la mortalité pendant toute l'étude.
75,2 % des patients étaient de sexe féminin. Les patients étaient majoritairement de type caucasien (67 %), asiatique (25 %) ou avaient la peau noire (6 %). L'âge moyen (écart type [ET], min.-max.) s'élevait à 54,0 (12,2, 20-79) ans. En tout, 51,9 % des patients souffraient d'une sclérose systémique cutanée diffuse (ScS) et 48,1 % d'une ScS cutanée limitée. La durée moyenne (ET) depuis la première apparition d'un symptôme non Raynaud était de 3,49 (1,7) ans. 49,0 % des patients ont reçu un traitement stable par mycophénolate et la profil de sécurité observé chez les patients sous mycophénolate ou non était similaire.
Déclin annuel de la CVF
Le déclin annuel de la CVF (en ml) sur 52 semaines a nettement diminué de 41,0 ml chez les patients sous Ofev par rapport au placebo (voir Tableau 3), ce qui correspond à un effet relatif du traitement de 43,8 %.
Tableau 3: Déclin annuel de la CVF (ml) sur 52 semaines
Placebo | Ofev 150 mg deux fois par jour | |
|---|---|---|
Nombre de patients évalués | 288 | 287 |
Taux1 (ET) du déclin sur 52 semaines | -93,3 (13,5) | -52,4 (13,8) |
Comparaison versus placebo | ||
Différence1 | 41,0 | |
IC à 95 % | (2,9, 79,0) | |
Valeur p | <0,05 | |
1 Sur la base d'une régression avec des coefficients aléatoires, ajustés pour le traitement, le sexe, la taille, l'âge, le statut ATA, la valeur initiale de CVF, CVF*la durée et traitement*la durée | ||
Une analyse exploratoire des données jusqu'à 100 semaines (durée de traitement max. dans l'étude SENSCIS) a montré que l'efficacité, avec un traitement par Ofev, sur le ralentissement de la progression de la PID-ScS durait plus de 52 semaines.
Dans deux analyses d'efficacité de sous-groupes prédéfinis, la différence moyenne de traitement relative au déclin de la CVF à la semaine 52 chez les patients selon la région et l'utilisation du mycophénolate a été étudiée (voir Figure 5).
Figure 5: Analyse de sous-groupes de la différence moyenne de traitement relative au déclin de la CVF (ml) à la semaine 52 selon la région et l'utilisation du mycophénolate (SENSCIS)
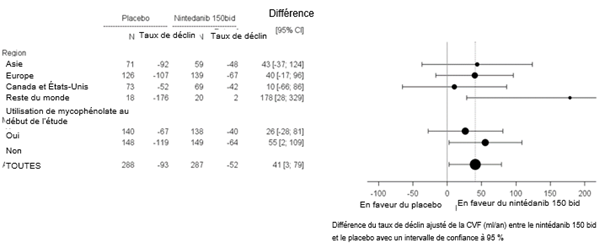
Variation en pourcentage de la capacité vitale forcée à partir du début de l'étude
La Figure 6 représente pour l'étude SENSCIS la variation en pourcentage de la CVF en ml à la semaine 52 par rapport à la valeur initiale. La dégradation de la fonction pulmonaire était moins importante chez la majorité des patients sous Ofev que chez les patients sous placebo.
Figure 6: Histogramme de la variation en pourcentage de la CVF (ml) à la semaine 52 par rapport à la valeur initiale selon le traitement et augmentation ou diminution en pourcentage de 5 (SENSCIS)a

Figure 7: Variation moyenne observée de la CVF par rapport à la valeur initiale (ml) sur 52 semaines

Tableau 4 Déclin annuel de la CVF (% de la valeur théorique) sur 52 semaines
Placebo | Ofev 150 mg deux fois par jour | |
|---|---|---|
Nombre de patients évalués | 288 | 287 |
Taux1 (ET) de déclin sur 52 semaines | -2,6 (0,4) | -1,4 (0,4) |
Compraison versus placebo | ||
Différence1 | 1,15 | |
IC à 95% | (0,09, 2,21) | |
Valeur p | <0,05 | |
1 Sur la base d'une régression avec des coefficients aléatoires et les effets de catégories fixes du traitement, le statut ATA, les effets continus fixes du temps, la valeur initiale de la CVF [% de la valeur théorique] y compris l'interaction entre le traitement et le temps et l'interaction entre la valeur initiale et le temps. L'intercept et le temps étaient donnés de manière spécifique au patient en tant qu'effet aléatoire. La variance d'erreur de mesure (within-patient error) inhérente à un patient a été modélisée au moyen d'une matrice de variance-covariance non structurée. La variabilité inter-individuelle a été modélisée au moyen d'une matrice de variance-covariance des composantes de variance. | ||
Variation du score de Rodnan Skin modifié (mRSS) à 52 semaines par rapport à la valeur initiale
La variation absolue moyenne ajustée du mRSS à 52 semaines par rapport à la valeur initiale était similaire entre le groupe sous Ofev (-2,17 (IC à 95% -2,69, -1,65)) et le groupe sous placebo (-1,96 (IC à 95% -2,48, -1,45)). La différence moyenne ajustée entre les groupes de traitement s'élevait à -0,21 (IC à 95% -0,94, 0,53; p = 0,5785).
Variation du score total au Saint George's Respiratory Questionnaire (SGRQ) à 52 semaines par rapport à la valeur initiale
La variation absolue moyenne ajustée du score total de SGRD à 52 semaines par rapport à la valeur initiale était similaire entre le groupe sous Ofev (0,81 (IC à 95% -0,92, 2,55)) et le groupe sous placebo (-0,88 (IC à 95% -2,58, 0,82)). La différence moyenne ajustée entre les groupes de traitement s'élevait à 1,69 (IC à 95% -0,73, 4,12; p = 0,1711).
Analyse de survie
Sur toute l'étude, la mortalité entre le groupe Ofev (N = 10; 3,5 %) et le groupe sous placebo (N = 9; 3,1 %) était similaire. Sur toute l'étude, l'analyse du temps jusqu'au décès a donné un HR de 1,16 (IC à 95% 0,47, 2,84; p = 0,7535).
Effet sur l'intervalle QT
Dans une étude spécifique auprès de patients atteints d'un cancer du rein à cellules claires, des mesures du QT/QTc ont révélé que ni l'administration d'une dose orale unique de 200 mg de nintédanib, ni l'administration de doses orales répétées de 200 mg de nintédanib deux fois par jour pendant 15 jours n'a prolongé l'intervalle QTcF. Aucune étude approfondie complète de l'intervalle QT n'a été effectuée sur le nintédanib.
Études auprès de la population pédiatrique
Aucune étude clinique n'a été effectuée auprès d'enfants et d'adolescents.
Pharmacocinétique
Les propriétés pharmacocinétiques du nintédanib étaient similaires chez des volontaires sains, des patients atteints de FPI, de PID-ScS, de PID et des patients atteints de cancer.
La pharmacocinétique du nintédanib est linéaire par rapport au temps. Une proportionnalité entre la dose et l'augmentation de l'exposition au nintédanib a été démontrée à l'aide de doses croissantes (allant de 50 à 450 mg une fois par jour et de 150 à 300 mg deux fois par jour). Après l'administration de doses multiples chez des patients atteints de FPI, l'accumulation se traduisait par une AUCτ augmentée d'un facteur 1,76. Les concentrations plasmatiques à l'état d'équilibre étaient atteintes après une semaine d'administration. Les concentrations minimales de nintédanib sont restées stables pendant plus d'un an. La pharmacocinétique du nintédanib présente une variabilité interindividuelle modérée à élevée (coefficient de variation de 30 % à 70 % pour les paramètres pharmacocinétiques standard). La variabilité intra-individuelle est faible à modérée (coefficient de variation inférieur à 40 %).
Absorption
Après l'administration orale de nintédanib en capsules molles de gélatine avec un repas, les concentrations plasmatiques maximales de nintédanib ont été atteintes en l'espace d'environ 2 à 4 heures (entre 0,5 et 8 heures). La biodisponibilité absolue d'une dose de 100 mg chez des volontaires sains était de 4,69 % (IC à 90 %: 3,62-6,08). L'absorption et la biodisponibilité sont réduites par l'influence de transporteurs (P-gp), par un métabolisme substantiel de premier passage et éventuellement par la faible solubilité du nintédanib dans un milieu de pH neutre.
Après l'ingestion de nourriture, l'exposition au nintédanib était accrue d'environ 20 % par rapport à une prise à jeun (IC: 95,3 %;152,5 %) et l'absorption était ralentie (tmax médian: 2,00 heures à jeun, 3,98 h après une prise de nourriture).
Distribution
Le nintédanib présente une cinétique d'élimination au moins biphasique. Un volume de distribution élevé (Vss: 1050 l) a été observé après une administration par perfusion intraveineuse.
La liaison du nintédanib aux protéines plasmatiques humaines était élevée in vitro (97,8 %). On suppose que l'albumine sérique est la principale protéine de liaison. Le nintédanib est principalement distribué dans le plasma, avec un rapport sang-plasma de 0,869.
Métabolisme
La métabolisation du nintédanib s'effectue essentiellement par une hydrolyse due à des estérases. Il en résulte la formation du métabolite principal, le groupement acide libre BIBF 1202. Des enzymes (UGT 1A1, UGT 1A7, UGT 1A8 et UGT 1A10) transforment ensuite le BIBF 1202 en glucuronide BIBF 1202.
La biodégradation du nintédanib ne s'effectue qu'en faible mesure par voie des enzymes du CYP (surtout CYP 3A4). Dans l'étude ADME, la plus grande partie de la radioactivité retrouvée dans le plasma humain était répartie entre le nintédanib (24 %), le BIBF 1202 (32 %) et le glucuronide BIBF 1202 (30 %), tandis que les principaux métabolites CYP-dépendants étaient indétectables dans le plasma humain. In vitro, le métabolisme CYP-dépendant correspondait à environ 5 %, par rapport à environ 25 % pour l'hydrolyse de l'ester dans ces conditions expérimentales.
Élimination
La clairance plasmatique totale après une perfusion intraveineuse était élevée (CL: 1390 ml/min). L'élimination de la substance active inchangée dans les urines en l'espace de 48 h était d'environ 0,05 % après l'administration d'une dose orale et d'environ 1,4 % après l'administration d'une dose intraveineuse. La clairance rénale était de 20 ml/min.
Excrétion
Après administration orale de nintédanib radiomarqué au [14C], la voie principale d'élimination de la radioactivité liée au médicament était l'excrétion fécale/biliaire (93,4 % de la dose). La plus grande partie de la dose d'Ofev (58 % de la dose) était éliminée dans les selles sous forme de BIBF 1202; 20 % étaient éliminés en tant que substance inchangée et moins de 7 % sous forme de divers métabolites de moindre importance. La contribution de l'excrétion rénale à la clairance totale était faible (0,649 % de la dose). La dose récupérée était considérée comme totale (plus de 90 %) dans les 4 jours suivant l'administration. La demi-vie terminale du nintédanib était de 10 à 15 h.
Rapport exposition/réponse
Des analyses exploratoires pour évaluer la relation entre la pharmacocinétique et les effets indésirables, effectuées à partir des données de l'étude de phase II sur la FPI, ont montré qu'une exposition supérieure au nintédanib était en tendance associée à une augmentation des taux d'enzymes hépatiques (voir «Mises en garde et précautions»).
Cinétique pour certains groupes de patients, facteurs intrinsèques et extrinsèques
D'après les résultats d'une analyse pharmacocinétique de population portant sur des patients atteints de FPI et de cancer bronchique non à petites cellules (CBNPC) (N = 1191) et d'après des études descriptives, l'exposition au nintédanib n'était pas influencée par le sexe (après correction en fonction du poids corporel), l'insuffisance rénale légère à modérée (estimée sur la base de la clairance de la créatinine), la consommation d'alcool ou le génotype de la P-gp. L'analyse pharmacocinétique de population a révélé que l'exposition au nintédanib était modérément influencée par l'âge, le poids corporel et l'origine ethnique. Ces facteurs sont présentés plus en détail ci-dessous. Dans le contexte de la forte variabilité inter-individuelle observée pour l'exposition, les effets modérés de ces co-variables ne permettent pas de donner des recommandations d'ajustement posologique (voir «Mises en garde et précautions»).
Âge
L'exposition au nintédanib augmente de façon linéaire avec l'âge. L'AUCτ,ss d'un patient de 45 ans (5e percentile) était inférieure de 16 % et celle d'un un patient de 76 ans (95e percentile) était supérieure de 13 % à celle d'un patient âgé de 62 ans (âge médian). La tranche d'âge couverte par l'analyse allait de 29 à 85 ans et environ 5 % de la population avaient plus de 75 ans.
Aucune étude n'a été effectuée auprès d'enfants et d'adolescents.
Poids corporel
Une corrélation inverse a été observée entre le poids corporel et l'exposition au nintédanib. L'AUCτ,ss d'un patient de 50 kg (5e percentile) était supérieure de 25 % et celle d'un un patient de 100 kg (95e percentile) était inférieure de 19 % à celle d'un patient pesant 71,5 kg (poids corporel médian).
Origine ethnique
La moyenne de la population d'exposition au nintédanib était supérieure de 33 à 50 % chez les Chinois, Taïwanais et Indiens et supérieure de 16 % chez les patients japonais, et plus faible de 16 à 22 % chez les Coréens, comparé aux patients caucasoïdes (après correction en fonction du poids corporel). Les données obtenues chez les patients noirs sont très limitées mais du même ordre que celles observées chez les patients caucasoïdes.
Fumeurs
Dans l'analyse pharmacocinétique de population, l'exposition au nintédanib était plus faible de 21 % chez les fumeurs actuels que chez les ex-fumeurs et les non-fumeurs de toujours. L'ampleur de cet effet ne justifie pas un ajustement posologique.
Insuffisance hépatique
Dans une étude dédiée de Phase I en dose unique, l'exposition systémique au nintédanib évaluée par la Cmax et l'ASC a été 2,2 fois plus élevée chez les sujets présentant une insuffisance hépatique légère (Child Pugh A; Cmax : IC à 90 %: 1,3 – 3,7et ASC: 1,2 – 3,8) que chez les volontaires sains. Chez les sujets présentant une insuffisance hépatique modérée (Child Pugh B), l'exposition systémique a été augmentée de 7,6 fois selon la Cmax (IC à 90 %: 4,4 – 13,2) et 8,7 fois selon l'ASC (IC à 90 %: 5,7 – 13,1) comparativement aux volontaires sains. Les sujets présentant une insuffisance hépatique sévère (Child Pugh C) n'ont pas été étudiés.
Insuffisance rénale
La pharmacocinétique du nintédanib n'a pas été examinée dans une étude PC spécifique chez les patients présentant une insuffisance rénale. D'après les résultats de l'analyse pharmacocinétique de population, une insuffisance rénale légère (ClCr de 60 à 90 ml/min) ou modérée (ClCr de 30 à 60 ml/min) n'a pas influencé l'exposition au nintédanib. Les données disponibles sur les patients présentant une insuffisance rénale sévère (ClCr inférieure à 30 ml/min) sont limitées.
Données précliniques
Toxicologie générale
Les études de toxicité après administration en dose unique chez le rat et la souris indiquent un faible potentiel de toxicité aiguë du nintédanib. Dans des études sur la toxicité en administration répétée chez le rat, les effets indésirables (tels que épaississement des plaques épiphysaires, lésions des incisives) étaient principalement liés au mécanisme d'action du nintédanib (inhibition du VEGFR-2). Des anomalies de ce type sont connues pour d'autres inhibiteurs du VEGFR-2 et peuvent être considérées comme des effets de classe.
Des diarrhées et des vomissements, associés à une moindre consommation de nourriture et à une perte de poids, ont été observés dans des études de toxicité avec administration de doses multiples chez d'autres animaux que les rongeurs.
Aucun indice d'augmentation des taux d'enzymes hépatiques n'a été trouvé chez le rat, le chien ou le singe cynomolgus. Uniquement chez le singe Rhésus, de légères élévations des enzymes hépatiques non attribuables à des effets indésirables sérieux tels que la diarrhée ont été observées.
Mutagénicité/carcinogénicité
Les études de génotoxicité n'ont révélé aucun potentiel mutagène du nintédanib.
Les études de 2 ans sur la carcinogénicité chez la souris et le rat n'ont pas mis en évidence de potentiel carcinogène du nintédanib.
Toxicité de reproduction
Chez le rat, une létalité embryo-fœtale et des effets tératogènes ont été observés à des niveaux d'exposition environ 3,6 à 7,2 fois inférieurs à l'exposition humaine observée à la dose quotidienne maximale recommandée chez l'homme (DMRH), de 150 mg deux fois par jour. Des effets sur le développement du squelette axial et des grosses artères ont également été observés à des niveaux d'exposition environ 12 à 18 fois inférieurs à la DMRH.
Chez le lapin, une létalité embryo-fœtale et des effets tératogènes ont été observés à une exposition environ 3 fois supérieure à la DMRH, tandis qu'une plus faible exposition correspondant à la DMRH de 150 mg deux fois par jour était déjà associée à des effets plus équivoques sur le développement embryo-fœtal du squelette axial et du cœur.
Une étude sur la fertilité du rat mâle et le développement embryonnaire précoce jusqu'à l'implantation n'a révélé aucune influence sur le taux de reproduction des mâles ou sur leur fertilité à des expositions environ 3 fois supérieures à la DMRH.
Chez des rates, de faibles quantités de nintédanib et/ou de ses métabolites radiomarqués ont été excrétées dans le lait (≤0,5 % de la dose administrée).
Remarques particulières
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Tenir hors de portée des enfants.
Conserver à température ambiante (15-25°C). Conserver le récipient dans son emballage d'origine pour protéger son contenu de l'humidité.
Numéro d’autorisation
65330 (Swissmedic)
Titulaire de l’autorisation
Boehringer Ingelheim (Schweiz) GmbH, Bâle.
Mise à jour de l’information
Octobre 2020
Reviews (0)

Free consultation with an experienced pharmacist
Describe the symptoms or the right drug - we will help you choose its dosage or analogue, place an order with home delivery or just consult.
We are 14 pharmacists and 0 bots. We will always be in touch with you and will be able to communicate at any time.
Bestsellers

Milupa Aptamil Pepti Syneo Can 400g
Product code: 7748334Aptamil Pepti Syneo is a special food for babies from birth who are allergic to cow's milk protein a..
55.08 CHF


Weleda Baby Calendula Wind- und Wetterbalsam 30ml
Product code: 5455461Der Weleda Baby Calendula Wind- und Wetterbalsam schützt die sensible Babyhaut intensiv bei Kälte un..
15.35 CHF


HerpoTherm herpes pen
Product code: 7798882Herpotherm® - the heating pen Cold sores are not only unattractive but can also be very painful. Th..
78.48 CHF





