




Forxiga Filmtabl 5 mg 98 pcs
Forxiga Filmtabl 5 mg 98 Stk
-
300.96 CHF
- Price in reward points: 3131

- Availability: In stock
- Brand: ASTRAZENECA AG
- Product Code: 6143108
- ATC-code A10BK01
- EAN 7680651760022
Ingredients:
Magnesium stearat, Lactose-1-Wasser 25 mg, Polyvinylalkohol, Titandioxid (E171), Talkum, Eisen(III)-oxid (E172), Siliciumdioxid anhydrat, Crospovidon, Macrogol 3350, Cellulose, mikrokristalline, Dapagliflozin 5 mg , Dapagliflozin-[(2S)-Propan-1,2-diol] (1:1)-1-Wasser 6.15 mg, Überzug:.

Variants

Forxiga Filmtabletten 5mg 28 Stück
113.05 CHF
Description
 Deutsch
Deutsch French
French Italian
ItalianWas ist Forxiga und wann wird es angewendet?
Auf Verschreibung des Arztes oder der Ärztin.
Forxiga enthält den Wirkstoff Dapagliflozin. Dieser gehört zu einer Arzneimittelgruppe der sogenannten SGLT2-Hemmer. Diese blockieren das SGLT2 Protein in den Nieren. Dadurch wird Zucker (Glucose), Salz (Natrium) und Wasser über den Urin aus dem Körper ausgeschieden.
Diabetes
Dapagliflozin führt zu einer Zuckerausscheidung über den Urin und senkt dadurch den Blutzuckerspiegel bei erwachsenen Patienten mit Diabetes Typ 2.
Forxiga wurde Ihnen verschrieben, da Ihr Blutzucker durch Diät und körperliche Betätigung nicht ausreichend gesenkt werden kann. In Kombination mit einer Diät und ausreichender Bewegung kann Forxiga alleine oder in Kombination mit anderen Antidiabetika eingenommen werden.
Herzinsuffizienz
Forxiga wurde Ihnen zur Ergänzung der Behandlung Ihrer Herzinsuffizienz verschrieben.
Was sollte dazu beachtet werden?
Was ist Diabetes Typ 2?
Diabetes Typ 2 ist eine Erkrankung, bei welcher der Körper nicht genügend Insulin bildet und das körpereigene Insulin nicht so stark wirkt wie es sollte. Ausserdem bildet die Leber bei vielen Diabetes-Erkrankten zu viel Zucker. Dadurch reichert sich Zucker im Blut an, was zu schwerwiegenden gesundheitlichen Problemen führen kann.
Das Hauptziel der Diabetes-Behandlung besteht darin, den Blutzucker auf einen normalen Wert zu senken. Durch die Senkung und Kontrolle des Blutzuckerspiegels lassen sich Diabetes-Komplikationen wie Herzerkrankungen, Nierenschädigung, Erblindung und Amputationen hinauszögern oder verhindern.
Für die Einstellung Ihres Diabetes sind Diät und Bewegung auch dann erforderlich, wenn Sie Arzneimittel einnehmen. Daher ist es wichtig, im Hinblick auf Diät und Bewegung den Rat Ihres Arztes oder Ihrer Ärztin zu befolgen.
Lassen Sie sich von Ihrem Arzt bzw. Ihrer Ärztin erklären, wie Sie einen zu niedrigen (Hypoglykämie) oder zu hohen (Hyperglykämie) Blutzuckerspiegel und andere mögliche Diabetes-Komplikationen verhindern, erkennen und behandeln können.
Ihr Arzt bzw. Ihre Ärztin wird Ihren Diabetes mit Hilfe regelmässiger Blutuntersuchungen überwachen, wozu Messungen der Glukose im Blut und des Hämoglobin-A1c-Wertes (HbA1c) gehören.
Sie sollten Ihren Blutzuckerspiegel regelmässig kontrollieren, damit Sie selbst und Ihr Arzt bzw. Ihre Ärztin den Verlauf überwachen können.
Was ist eine Herzinsuffizienz?
Bei einer Herzinsuffizienz ist die Pumpleistung des Herzmuskels nicht mehr stark genug, um die Lunge und den restlichen Körper ausreichend mit Blut zu versorgen. Dies kann zu schwerwiegenden medizinischen Problemen führen und einen Krankenhausaufenthalt nötig machen. Die häufigsten Symptome einer Herzinsuffizienz sind Kurzatmigkeit, Erschöpfung, Müdigkeit und ein Anschwellen der Knöchel. Die Behandlung der Herzinsuffizienz dient hauptsächlich dazu den Krankheitsverlauf zu verlangsamen und die Symptome zu reduzieren. Die Notwendigkeit von Krankenhausaufenthalten kann dadurch sinken.
Zucker im Harn
Wegen der Wirkungsweise von Forxiga ist in Ihrem Urin Zucker nachweisbar, solange sie Forxiga einnehmen.
Wann darf Forxiga nicht eingenommen werden?
Forxiga darf nicht angewendet werden bei bekannter Überempfindlichkeit auf den Wirkstoff oder einen der Hilfsstoffe.
Wann ist bei der Einnahme von Forxiga Vorsicht geboten?
Sprechen Sie mit Ihrem Arzt bzw. mit Ihrer Ärztin, bevor Sie Forxiga einnehmen, insbesondere:
- wenn Sie an Diabetes Typ 1 erkrankt sind.
- wenn bei Diabetikern das Risiko einer diabetischen Ketoazidose besteht (gefährlich hohe Konzentrationen bestimmter Säuren, sogenannter Ketonen, im Blut oder Urin). Risikofaktoren für das Auftreten einer Ketoazidose sind zum Beispiel Erkrankungen der Bauchspeicheldrüse, kohlenhydratarme Ernährung, verminderte Kalorienaufnahme, Änderungen des Insulinbedarfs sowie Alkoholismus. Es handelt sich dabei um eine schwerwiegende Stoffwechselentgleisung, die auch tödlich verlaufen kann, wenn sie nicht unverzüglich behandelt wird. Beim Auftreten von Symptomen wie Übelkeit, Erbrechen, Appetitlosigkeit, Bauchschmerzen, übermässigem Durst, Atemschwierigkeiten, Verwirrtheit, ungewöhnliche Erschöpfung oder Müdigkeit konsultieren Sie sofort Ihren Arzt oder Ihre Ärztin und beenden Sie die Einnahme von Forxiga umgehend.
- wenn Sie eine Nierenfunktionsstörung haben.
- wenn Sie eine Leberfunktionsstörung haben.
- wenn Sie unter Pilzinfektionen im Intimbereich leiden oder je gelitten haben.
- wenn Sie Arzneimittel einnehmen, welche Ihr Immunsystem schwächen.
Wenn einer der oben genannten Punkte auf Sie zutrifft oder wenn Sie sich nicht ganz sicher sind, sprechen Sie mit Ihrem Arzt oder Apotheker bzw. Ihrer Ärztin oder Apothekerin, bevor Sie Forxiga einnehmen.
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wenn Sie andere Arzneimittel einnehmen bzw. vor kurzem eingenommen haben, auch wenn es sich um nicht verschreibungspflichtige Arzneimittel oder pflanzliche Präparate handelt.
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin insbesondere dann,
- wenn Sie mit Arzneimitteln zur Blutdrucksenkung behandelt werden oder bereits früher einen zu niedrigen Blutdruck (Hypotonie) hatten.
- wenn Sie andere Arzneimittel zur Senkung des Blutzuckerspiegels einnehmen/anwenden. Ihr Arzt wird möglicherweise die Dosis dieser anderen Arzneimittel anpassen.
Wenn bei Ihnen während der Einnahme von Forxiga Symptome wie Schmerzen oder Druckempfindlichkeit, Hautrötung, Schwellungen im Bereich der Genitalien und des Dammes, Fieber oder Unwohlsein auftreten, beenden Sie die Einnahme von Forxiga und setzen Sie sich umgehend mit Ihrem Arzt oder Ihrer Ärztin oder dem nächstgelegenen Krankenhaus in Verbindung. Diese Symptome können Anzeichen für eine seltene, aber lebensbedrohliche bakterielle Infektion sein, die das Gewebe unter der Haut zerstört (auch als Fournier-Gangrän bezeichnet) und sofort behandelt werden muss.
Falls Ihnen während der Behandlung mit Forxiga schwindlig ist, verzichten Sie auf das Steuern eines Fahrzeugs sowie auf das Bedienen von Werkzeugen oder Maschinen.
Forxiga wurde bei Kindern und Jugendlichen unter 18 Jahren nicht untersucht. Daher sollte Forxiga von Kindern und Jugendlichen nicht eingenommen werden.
Forxiga enthält Lactose (Milchzucker). Bitte nehmen Sie Forxiga erst nach Rücksprache mit Ihrem Arzt bzw. Ihrer Ärztin ein, wenn Ihnen bekannt ist, dass Sie unter einer Zuckerunverträglichkeit leiden.
Informieren Sie Ihren Arzt, Apotheker bzw. Ihre Ärztin, Apothekerin, wenn Sie
- an anderen Krankheiten leiden,
- Allergien haben oder
- andere Arzneimittel (auch selbst gekaufte!) einnehmen!
Darf Forxiga während einer Schwangerschaft oder in der Stillzeit eingenommen werden?
Wenn Sie schwanger sind, wenn Sie glauben schwanger zu sein oder eine Schwangerschaft planen, fragen Sie Ihren Arzt bzw. Ihre Ärztin vor der Einnahme dieses Arzneimittels um Rat.
Informieren Sie Ihren Arzt bzw. Ihre Ärztin vor der Einnahme von Forxiga, wenn Sie stillen oder stillen möchten. Wenden Sie Forxiga nicht an, wenn Sie stillen.
Wie verwenden Sie Forxiga?
Diabetes
Die empfohlene Anfangsdosis für die Behandlung des Typ 2 Diabetes ist Forxiga 5 mg 1x täglich. Bei Bedarf kann der Arzt bzw. die Ärztin die Dosis auf Forxiga 10 mg 1x täglich erhöhen.
Herzinsufizienz
Die empfohlene Dosis für die Behandlung der Herzinsuffizienz ist Forxiga 10 mg 1x täglich.
Schlucken Sie die Tablette als Ganzes mit etwas Wasser. Sie können die Tabletten unabhängig von einer Mahlzeit einnehmen.
Die Tabletteneinnahme kann zu jeder Tageszeit erfolgen. Versuchen Sie dennoch, die Tabletten jeden Tag etwa zur gleichen Zeit einzunehmen. Dadurch denken Sie leichter daran, Ihre Tabletten zu nehmen. Nehmen Sie Forxiga immer genau nach Anweisung Ihres Arztes bzw. Ihrer Ärztin ein. Brechen Sie die Einnahme von Forxiga nicht ohne vorherige Rücksprache mit Ihrem Arzt bzw. Ihrer Ärztin ab.
Sollten Sie die Einnahme von Forxiga einmal vergessen haben, nehmen Sie das Arzneimittel ein, sobald Sie sich daran erinnern. Wenn Sie es erst am nächsten Tag bemerken, lassen Sie die vergessene Dosis aus und fahren Sie einfach mit Ihrem gewohnten Einnahmeschema fort. Nehmen Sie nicht die doppelte Dosis ein. Wenn Sie mehr Forxiga Tabletten eingenommen haben, als Sie sollten, wenden Sie sich umgehend an Ihren Arzt bzw. Ihre Ärztin oder begeben Sie sich in ein Krankenhaus. Wenden Sie sich an Ihren Apotheker bzw. Ihre Apothekerin, wenn Sie Fragen im Zusammenhang mit einer ausgelassenen Dosis haben.
Die Anwendung und Sicherheit von Forxiga bei Kindern und Jugendlichen / bei Kindern unter 18 Jahren ist bisher nicht geprüft worden.
Ändern Sie nicht von sich aus die verschriebene Dosierung. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. mit Ihrer Ärztin oder Apothekerin.
Welche Nebenwirkungen kann Forxiga haben?
Während der Behandlung mit Forxiga können die folgenden Nebenwirkungen auftreten:
Sehr häufig (betrifft mehr als einen von 10 Anwendern)
Niedriger Blutzuckerspiegel (Hypoglykämie) in Patienten mit Diabetes ‑ in Kombination mit andern Arzneimitteln gegen erhöhten Blutzucker vom Typ Sulfonylharnstoff oder Insulin.
Häufig (betrifft 1 bis 10 von 100 Anwendern)
Pilzinfektion (Candidose) des Penis oder der Vagina; Harnwegsinfektionen; Ausscheidung von mehr Urin oder häufigeres Wasserlassen; Rückenschmerzen; mehr rote Blutkörperchen im Blut (erhöhter Hämatokrit), wird bei Blutuntersuchungen festgestellt; Verlust von zu viel Körperwasser (Dehydrierung). Mögliche Anzeichen sind sehr trockener oder klebriger Mund, starkes Durstgefühl, starkes Schläfrigkeits- oder Müdigkeitsgefühl, verminderte oder fehlende Urinausscheidung, beschleunigter Herzschlag.
Gelegentlich (betrifft 1 bis 10 von 1'000 Anwendern)
Ungewöhnliche Vaginalblutungen, Ausfluss, Juckreiz oder Geruch; Durst; Verstopfung; übermässiges Schwitzen; Aufwachen in der Nacht wegen Harndrangs; erschwerte Blasenentleerung; Veränderungen im Rahmen von Laboruntersuchungen des Blutes (zum Beispiel Harnstoff).
Selten (betrifft 1 bis 10 von 10'000 Anwendern)
Diabetische Ketoazidose.
Sehr selten (betrifft weniger als 1 von 10'000 Anwendern)
Nekrotisierende Fasziitis des Perineums (Fournier-Gangrän).
Setzen Sie Forxiga ab, und wenden Sie sich schnellstmöglich an Ihren Arzt oder Ihre Ärztin, wenn Sie eine der folgenden schwerwiegenden Nebenwirkungen bemerken:
- Harnwegsinfektion. Anzeichen einer schweren Harnwegsinfektion sind: Fieber und/oder Schüttelfrost, brennendes Gefühl beim Wasserlassen, Schmerzen im Rücken oder an der Seite.
- Überempfindlichkeitsreaktionen (wie zum Beispiel Schwellung der Haut oder Schleimhaut, Nesselsucht).
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt, Apotheker bzw. Ihre Ärztin, Apothekerin. Dies gilt insbesondere auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Was ist ferner zu beachten?
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Lagerungshinweis
Nicht über 30 °C lagern.
In der Originalverpackung aufbewahren.
Ausser Reichweite von Kindern aufbewahren.
Weitere Hinweise
Weitere Auskünfte erteilt Ihnen Ihr Arzt, Apotheker bzw. Ihre Ärztin, Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in Forxiga enthalten?
Wirkstoffe
1 Filmtablette Forxiga 5 mg enthält 5 mg Dapagliflozin als Dapagliflozin-Monohydrat-Propandiol.
1 Filmtablette Forxiga 10 mg enthält 10 mg Dapagliflozin als Dapagliflozin-Monohydrat-Propandiol.
Hilfsstoffe
Tablettenkern
Mikrokristalline Cellulose (E460i)
Lactose (25 mg Lactose in 5 mg Filmtabletten. 50 mg Lactose in 10 mg Filmtabletten)
Crospovidon (E1202)
Siliciumdioxid (E551)
Magnesiumstearat (E470b)
Filmüberzug
Poly(vinylalkohol) (E1203)
Titandioxid (E171)
Macrogol 3350
Talkum (E553b)
Gelbes Eisenoxid (E172)
Zulassungsnummer
65176 (Swissmedic)
Wo erhalten Sie Forxiga? Welche Packungen sind erhältlich?
In Apotheken nur gegen ärztliche Verschreibung.
Packungen zu 28 und 98 Filmtabletten à 5 mg.
Packungen zu 28 und 98 Filmtabletten à 10 mg.
Zulassungsinhaberin
AstraZeneca AG, 6340 Baar
Diese Packungsbeilage wurde im Juli 2020 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
Qu’est-ce que Forxiga et quand doit-il être utilisé?
Selon prescription du médecin.
Forxiga contient le principe actif dapagliflozine. Cette substance appartient à un groupe de médicaments appelés inhibiteurs du SGLT2. Ces derniers inhibent la protéine SGLT2 dans les reins, ce qui permet d'éliminer le sucre (glucose), le sel (sodium) et l'eau de l'organisme par les urines.
Diabète
La dapagliflozine conduit à une élimination du sucre par les urines et fait baisser la glycémie (taux de sucre dans le sang) chez les patients adultes souffrant de diabète de type 2.
Forxiga vous a été prescrit, car le régime alimentaire et l'exercice physique ne suffisaient pas à abaisser suffisamment votre glycémie. Forxiga peut être pris seul ou en association avec d'autres antidiabétiques en combinaison avec un régime alimentaire et suffisamment d'exercice physique.
Insuffisance cardiaque
Forxiga vous a été prescrit en complément de votre traitement de l'insuffisance cardiaque.
De quoi faut-il tenir compte en dehors du traitement?
Qu'est-ce que le diabète de type 2 ?
Le diabète de type 2 est une maladie dans le cadre de laquelle le corps ne produit pas suffisamment d'insuline et l'effet de l'insuline produite par l'organisme n'est pas aussi puissant qu'il devrait l'être. En outre, le foie produit trop de sucre chez de nombreux patients atteints de diabète. Dans ces circonstances, le sucre s'accumule dans le sang, ce qui peut conduire à de graves problèmes de santé.
Le traitement antidiabétique a pour but principal de ramener la glycémie à des valeurs normales. La baisse et le contrôle de la glycémie permettent de retarder ou d'éviter les complications du diabète telles que maladies cardiaques, atteinte rénale, cécité et amputations.
Même si vous prenez des médicaments, un régime alimentaire et de l'exercice physique sont aussi nécessaires pour le contrôle du diabète. C'est pourquoi il est important que vous suiviez les conseils de votre médecin concernant le régime alimentaire et l'exercice physique.
Votre médecin vous expliquera comment éviter, reconnaître et traiter une hypoglycémie (taux de sucre trop bas dans le sang), une hyperglycémie (taux de sucre trop élevé dans le sang) et d'autres complications possibles du diabète.
Le médecin contrôlera l'évolution de votre diabète par des analyses de sang régulières telles que mesures de la glycémie (taux de sucre dans le sang) et de l'hémoglobine glyquée (HbA1c).
Vous devriez contrôler régulièrement votre glycémie afin de surveiller ses variations avec votre médecin.
Qu'est-ce que l'insuffisance cardiaque ?
En cas d'insuffisance cardiaque, le cœur ne parvient plus à pomper suffisamment de sang pour l'acheminer dans les poumons et le reste de l'organisme. Cela peut entraîner de graves problèmes de santé et nécessiter une hospitalisation. Les symptômes les plus fréquents de l'insuffisance cardiaque sont l'essoufflement, l'épuisement, la fatigue et le gonflement des chevilles. Le traitement de l'insuffisance cardiaque sert principalement à ralentir l'évolution de la maladie et à diminuer les symptômes, ce qui permet de réduire la nécessité d'hospitalisation.
Glucose urinaire
En raison du mode d'action de Forxiga, du sucre peut être détecté dans votre urine aussi longtemps que vous prenez Forxiga.
Quand Forxiga ne doit-il pas être pris?
Forxiga ne doit pas être utilisé en cas d'hypersensibilité connue au principe actif ou à l'un des excipients.
Quelles sont les précautions à observer lors de la prise de Forxiga?
Adressez-vous à votre médecin avant de prendre Forxiga, notamment dans les cas suivants:
- si vous avez un diabète de type 1;
- si chez des personnes diabétiques, un risque d'acidocétose diabétique est présent (concentrations dangereusement élevées de certains acides, corps cétoniques, dans l'urine ou le sang). Les facteurs de risque contribuant à la survenue d'une acidocétose sont par exemple des affections du pancréas, une alimentation pauvre en glucides, une diminution de l'apport calorique, changements des besoins en insuline ainsi que l'alcoolisme. Il s'agit d'un dérèglement métabolique sévère dont l'issue peut également être fatale s'il n'est pas traité immédiatement. Lors de la survenue de symptômes tels que nausées, vomissements, manque d'appétit, douleurs abdominales, soif excessive, difficultés respiratoires, confusion, fatigue ou épuisement inhabituels consultez immédiatement votre médecin et arrêtez immédiatement de prendre Forxiga.
- si vous avez des troubles de la fonction rénale;
- si vous avez des troubles de la fonction hépatique;
- si vous souffrez ou avez souffert de mycoses des parties intimes;
- si vous prenez des médicaments qui affaiblissent le système immunitaire.
Si l'une des situations mentionnées s'applique à votre situation ou en cas de doute, adressez-vous à votre médecin ou à votre pharmacien avant de prendre Forxiga.
Informez votre médecin ou votre pharmacien si vous prenez ou avez récemment pris tout autre médicament, même s'il s'agit de médicaments non soumis à ordonnance ou à base de plantes.
Avertissez votre médecin ou votre pharmacien notamment dans les cas suivants:
- si vous prenez des médicaments pour faire baisser la pression artérielle et avez un antécédent de pression artérielle trop basse (hypotension);
- si vous prenez ou utilisez d'autres traitements qui font baisser la glycémie. Votre médecin peut envisager de réduire la dose de ces autres médicaments.
Arrêtez de prendre Forxiga et contactez immédiatement votre médecin ou l'hôpital le plus proche si vous remarquez des symptômes comme des douleurs ou une sensibilité à la pression, une rougeur de la peau, des gonflements dans la zone des parties génitales et du périnée, une fièvre ou une sensation de malaise pendant la prise de Forxiga. Ces symptômes peuvent indiquer une infection bactérienne rare, quoique mettant la vie en danger, qui détruit le tissu sous-cutané (également appelée gangrène de Fournier) et qui doit être immédiatement traitée.
Ne conduisez pas ou n'utilisez pas d'outils ou de machines si vous avez des sensations de vertige lors du traitement par Forxiga.
Forxiga ne doit pas être administré chez les enfants et adolescents de moins de 18 ans, car il n'a pas fait l'objet d'études dans cette tranche d'âge.
Forxiga contient du lactose (sucre de lait). Si vous souffrez d'intolérance au sucre, ne prenez Forxiga qu'après en avoir parlé avec le médecin.
Veuillez informer votre médecin ou votre pharmacien si
- vous souffrez d'une autre maladie
- vous êtes allergique
- vous prenez déjà d'autres médicaments (même en automédication!).
Forxiga peut-il être pris pendant la grossesse ou l’allaitement?
Si vous êtes enceinte, si vous pensez l'être ou si vous planifiez une grossesse, demandez conseil à votre médecin avant de prendre ce médicament.
Si vous allaitez ou souhaitez allaiter, informez-en votre médecin avant de prendre Forxiga. N'utilisez pas Forxiga si vous allaitez.
Comment utiliser Forxiga?
Diabète
La dose initiale recommandée pour le traitement du diabète de type 2 est de 1 comprimé de Forxiga 5 mg 1x par jour. Si nécessaire, le médecin peut augmenter cette dose à 1 comprimé de Forxiga 10 mg 1x par jour.
Insuffisance cardiaque
La dose recommandée de Forxiga pour le traitement de l'insuffisance cardiaque est de 10 mg 1x par jour.
Avalez le comprimé entier avec un peu d'eau. Vous pouvez prendre les comprimés indépendamment des repas.
Vous pouvez prendre le comprimé à n'importe quel moment de la journée. Il est toutefois conseillé de le prendre chaque jour à peu près à la même heure. Vous éviterez ainsi de l'oublier. Veillez à toujours prendre Forxiga en suivant exactement les indications de votre médecin. N'interrompez pas le traitement par Forxiga sans en avoir parlé au préalable à votre médecin.
Si vous avez une fois oublié de prendre Forxiga, prenez le médicament dès que vous constatez l'oubli. Si vous ne remarquez votre oubli que le jour suivant, ne prenez pas la dose oubliée et poursuivez simplement selon votre schéma de prise habituel. Ne prenez pas de dose double. Si vous avez pris plus de comprimés de Forxiga que vous devriez, consultez immédiatement votre médecin ou rendez-vous à l'hôpital. Si vous avez des questions en lien avec l'oubli d'une dose, adressez-vous à votre pharmacien.
L'utilisation et la sécurité de Forxiga n'ont pas été établies à ce jour pour les enfants et adolescents / les enfants de moins de 18 ans.
Ne changez pas de votre propre chef le dosage prescrit. Adressez-vous à votre médecin ou à votre pharmacien si vous estimez que l'efficacité du médicament est trop faible ou au contraire trop forte.
Quels effets secondaires Forxiga peut-il provoquer?
Le traitement par Forxiga peut provoquer les effets secondaires suivants:
Très fréquent (concerne plus d'un utilisateur sur 10)
Taux de sucre bas dans le sang (hypoglycémie) chez les patients diabétiques, si ce médicament est pris en complément d'autres antidiabétiques de type sulfonylurée ou insuline.
Fréquent (concerne 1 à 10 utilisateurs sur 100)
Mycose (candidose) du pénis ou du vagin; infections urinaires; quantité d'urine plus importante que d'habitude ou besoin plus fréquent d'uriner; douleurs du dos; augmentation du taux de globules rouges dans le sang (hématocrite élevé), observée lors d'analyses de sang, perte d'une quantité trop importante d'eau corporelle (déshydratation). Les signes potentiels sont une bouche très sèche ou pâteuse, une soif excessive, une somnolence ou fatigue accrue, une miction réduite ou l'absence de miction, des pulsations cardiaques accélérées.
Occasionnel (concerne 1 à 10 utilisateurs sur 1000)
Saignements vaginaux inhabituels, écoulement, démangeaisons ou odeurs; soif; constipation; transpiration excessive; réveils nocturnes pour uriner; difficultés à uriner; modifications des analyses de sang (par exemple urée).
Rare (concerne 1 à 10 utilisateurs sur 10 000)
Acidocétose diabétique.
Très rare (concerne moins d'un utilisateur sur 10 000)
Fasciite nécrosante du périnée (gangrène de Fournier).
Interrompez la prise de Forxiga et contactez votre médecin dès que possible si vous remarquez l'un des effets indésirables suivants:
- infection urinaire. Les signes d'une infection urinaire grave sont: fièvre et/ou frissons, sensation de brûlure à la miction (lorsque vous urinez), douleurs dans le dos ou sur le flanc;
- réactions d'hypersensibilité (par exemple gonflement de la peau ou des muqueuses, urticaire).
Si vous remarquez des effets secondaires, veuillez en informer votre médecin ou votre pharmacien. Ceci vaut en particulier pour les effets secondaires non mentionnés dans cette notice d'emballage.
À quoi faut-il encore faire attention?
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques concernant le stockage
Ne pas conserver au-dessus de 30 °C.
Conserver dans l'emballage d'origine.
Conserver hors de la portée des enfants.
Remarques complémentaires
Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien, qui disposent d'une information détaillée destinée aux professionnels.
Que contient Forxiga?
Principes actifs
1 comprimé pelliculé de Forxiga 5 mg contient 5 mg de dapagliflozine sous forme de dapagliflozine propanediol monohydraté.
1 comprimé pelliculé de Forxiga 10 mg contient 10 mg de dapagliflozine sous forme de dapagliflozine propanediol monohydraté.
Excipients
Noyau du comprimé
Cellulose microcristalline (E460i)
Lactose (25 mg de lactose dans les comprimés pelliculés à 5 mg. 50 mg de lactose dans les comprimés pelliculés à 10 mg)
Crospovidone (E1202)
Dioxyde de silicium (E551)
Stéarate de magnésium (E470b)
Pelliculage
Poly(alcool vinylique) (E1203)
Dioxyde de titane (E171)
Macrogol 3350
Talc (E553b)
Oxyde de fer jaune (E172)
Numéro d’autorisation
65176 (Swissmedic)
Où obtenez-vous Forxiga? Quels sont les emballages à disposition sur le marché?
En pharmacie, sur ordonnance médicale.
Emballages de 28 et 98 comprimés pelliculés à 5 mg.
Emballages de 28 et 98 comprimés pelliculés à 10 mg.
Titulaire de l’autorisation
AstraZeneca AG, 6340 Baar
Cette notice d'emballage a été vérifiée pour la dernière fois en juillet 2020 par l'autorité de contrôle des médicaments (Swissmedic).
Che cos’è Forxiga e quando si usa?
Su prescrizione medica.
Forxiga contiene il principio attivo dapagliflozin. Dapagliflozin appartiene alla classe dei medicamenti noti come inibitori del SGLT2 che bloccano la proteina SGLT2 a livello renale. In tal modo lo zucchero (glucosio), il sale (sodio) e l'acqua vengono eliminati dall'organismo attraverso l'urina.
Diabete
Dapagliflozin induce l'escrezione di zucchero tramite l'urina determinando un abbassamento della glicemia (zucchero nel sangue) nei pazienti adulti con diabete di tipo 2.
Forxiga le è stato prescritto perché la dieta e il movimento non consentono, nel suo caso, di abbassare a sufficienza i suoi livelli glicemici. Insieme alla dieta e a una sufficiente attività fisica, Forxiga può essere assunto da solo o in combinazione con altri medicamenti antidiabetici.
Insufficienza cardiaca
Forxiga le è stato prescritto a integrazione del trattamento per l'insufficienza cardiaca.
Di che cosa occorre inoltre tener conto durante il trattamento?
Cos'è il diabete di tipo 2?
Il diabete di tipo 2 è una malattia che si manifesta quando l'organismo non produce insulina a sufficienza e questa insulina non ha un'azione sufficientemente forte. Inoltre, in molte persone che soffrono di diabete il fegato produce un eccesso di zucchero. In questi casi, lo zucchero si accumula nel sangue e ciò può causare gravi problemi di salute.
L'obiettivo primario del trattamento del diabete consiste nel riportare a livelli normali lo zucchero nel sangue. Riducendo e tenendo sotto controllo la glicemia è possibile ritardare o prevenire le complicanze del diabete quali malattie cardiache, danni renali, cecità e amputazioni.
Per tenere sotto controllo il diabete deve continuare la dieta prescritta e fare esercizio fisico anche se assume medicamenti. Per questo motivo è importante seguire i consigli del suo medico a riguardo.
Chieda al suo medico di spiegarle in che modo è possibile evitare, riconoscere e trattare l'ipoglicemia (livelli troppo bassi di zucchero nel sangue), l'iperglicemia (livelli troppo alti di zucchero nel sangue) e altre possibili complicanze del diabete.
Il suo medico monitorerà il suo diabete tramite regolari esami del sangue, incluse le misurazioni del glucosio nel sangue e del valore dell'emoglobina A1c (HbA1c).
Lei dovrebbe controllare regolarmente la sua glicemia affinché sia lei che il suo medico possiate seguirne l'andamento.
Che cos'è l'insufficienza cardiaca?
Nell'insufficienza cardiaca, la capacità del muscolo cardiaco di pompare sangue non è più sufficiente per fornire ai polmoni e al resto del corpo una quantità di sangue adeguata. Ciò può causare gravi problemi medici e rendere necessario un ricovero in ospedale. I sintomi più frequenti dell'insufficienza cardiaca sono affanno, spossatezza, stanchezza e gonfiore delle caviglie. Il trattamento dell'insufficienza cardiaca serve principalmente a rallentare il decorso della malattia e ad alleviare i sintomi. È possibile che in questo modo diminuisca la necessità di ricoveri in ospedale.
Zucchero nelle urine
Per via della modalità d'azione di Forxiga, per tutto il tempo che lei assume Forxiga, si evidenzia la presenza di zucchero nelle sue urine.
Quando non si può assumere Forxiga?
Forxiga non può essere usato in caso di ipersensibilità nota al principio attivo o a una delle sostanze ausiliarie.
Quando è richiesta prudenza nella somministrazione di Forxiga?
Consulti il suo medico prima di assumere Forxiga, in particolare:
- se soffre di diabete di tipo 1;
- se sussiste il rischio di chetoacidosi diabetica nelle persone diabetiche (concentrazione pericolosamente alta di determinati acidi, i cosiddetti corpi chetonici, nel sangue o nelle urine). I fattori di rischio per la comparsa di una chetoacidosi sono, ad esempio, malattie del pancreas, alimentazione povera di carboidrati, ridotta assunzione di calorie, variazioni nel fabbisogno di insulina nonché alcolismo. Si tratta di uno scompenso grave del metabolismo, che può avere anche conseguenze mortali, se non viene trattato immediatamente. Alla comparsa di sintomi quali nausea, vomito, mancanza di appetito, dolori addominali, sete eccessiva, difficoltà di respirazione, confusione, affaticamento o stanchezza insoliti, consulti immediatamente il suo medico e interrompa subito l'assunzione di Forxiga.
- se presenta disturbi della funzionalità renale;
- se presenta disturbi della funzionalità epatica;
- se ha o ha avuto in passato infezioni micotiche (da funghi) nelle parti intime;
- se fa uso di medicamenti che indeboliscono il sistema immunitario.
Se una di queste condizioni la riguarda, oppure non ne è del tutto sicuro, consulti il suo medico o il suo farmacista prima di assumere Forxiga.
Informi il suo medico o il suo farmacista se assume o ha assunto di recente altri medicamenti, anche se si tratta di medicamenti senza obbligo di prescrizione o di preparati vegetali.
Informi il suo medico o il suo farmacista soprattutto nel caso in cui:
- è in terapia con medicamenti per ridurre la pressione sanguigna o se ha già avuto in passato una pressione sanguigna troppo bassa (ipotensione);
- se assume/usa altri medicamenti per abbassare la glicemia. Il suo medico potrà modificare la dose di tali altri medicamenti.
Se durante l'assunzione di Forxiga manifesta sintomi come dolori o dolorabilità alla pressione, arrossamento cutaneo, gonfiori nella zona dei genitali e del perineo, febbre o malessere, interrompa l'assunzione di Forxiga e contatti immediatamente il suo medico o il più vicino ospedale. Questi sintomi possono essere segni di un'infezione batterica, rara ma potenzialmente letale, che distrugge il tessuto sotto pelle (condizione nota anche come gangrena di Fournier) e deve essere trattata immediatamente.
Se durante il trattamento con Forxiga manifesta vertigini, si astenga dal guidare veicoli o utilizzare attrezzi o macchinari.
L'uso di Forxiga nei bambini e negli adolescenti sotto i 18 anni finora non è stato esaminato. Pertanto Forxiga non deve essere assunto da bambini e adolescenti.
Forxiga contiene lattosio (zucchero del latte). Se sa di soffrire di un'intolleranza agli zuccheri, assuma Forxiga solo dopo aver consultato il suo medico.
Informi il suo medico o il suo farmacista, nel caso in cui
- soffre di altre malattie
- soffre di allergie o
- assume altri medicamenti (anche se acquistati di sua iniziativa)!
Si può assumere Forxiga durante la gravidanza o l’allattamento?
Se è incinta, presume di esserlo o sta pianificando una gravidanza, chieda consiglio al medico prima di assumere questo medicamento.
Prima di assumere Forxiga informi il medico se sta allattando o se desidera allattare. Non usi Forxiga se sta allattando.
Come usare Forxiga?
Diabete
La dose iniziale raccomandata per il trattamento del diabete di tipo 2 è 5 mg di Forxiga una volta al giorno. All'occorrenza il medico può aumentare la dose a 10 mg di Forxiga una volta al giorno.
Insufficienza cardiaca
La dose raccomandata per il trattamento dell'insufficienza cardiaca è 10 mg di Forxiga una volta al giorno.
Ingerisca la compressa intera con un po' d'acqua. Le compresse possono essere prese indipendentemente dai pasti, a qualsiasi ora. È tuttavia consigliabile assumere le compresse ogni giorno all'incirca alla stessa ora; in tal modo le sarà più facile ricordare di prenderle. Prenda Forxiga seguendo sempre esattamente le istruzioni del medico. Non ne interrompa l'assunzione senza aver dapprima consultato il medico.
Nel caso in cui dimenticasse di prendere Forxiga, assuma il medicamento non appena se ne ricorda. Se dovesse accorgersene solo il giorno seguente, non la assuma più e prosegua con il normale schema di assunzione. Non prenda una dose doppia. Se ha preso più compresse di Forxiga di quanto doveva, si rivolga immediatamente al suo medico oppure si rechi in ospedale. Se ha domande in merito alla mancata assunzione di una dose, si rivolga al suo farmacista.
L'uso e la sicurezza di Forxiga nei bambini e negli adolescenti / bambini sotto i 18 anni finora non sono stati esaminati.
Non modifichi di propria iniziativa la posologia prescritta. Se ritiene che l'azione del medicamento sia troppo debole o troppo forte ne parli al suo medico o al suo farmacista.
Quali effetti collaterali può avere Forxiga?
Durante il trattamento con Forxiga possono insorgere i seguenti effetti collaterali.
Molto comune (riguarda più di 1 utilizzatore su 10)
Basso livello di zuccheri nel sangue (ipoglicemia) in pazienti con diabete – in combinazione con altri medicamenti per trattare l'iperglicemia del tipo delle sulfoniluree o come l'insulina.
Comune (riguarda da 1 a 10 utilizzatori su 100)
Infezione micotica (candidosi) di pene o vagina; infezioni delle vie urinarie; aumento dell'emissione di urine o minzione più frequente; mal di schiena; aumento dei globuli rossi nel sangue (valori elevati di ematocrito) agli esami del sangue, perdita di una quantità eccessiva di liquidi corporei (disidratazione). I possibili segni sono bocca molto secca o appiccicosa, intensa sensazione di sete, forte sensazione di sonnolenza o stanchezza, diuresi ridotta o assente, battito cardiaco accelerato.
Non comune (riguarda da 1 a 10 utilizzatori su 1000)
Sanguinamento vaginale insolito, perdite, prurito o cattivo odore; sete; stitichezza; sudorazione eccessiva; risvegli notturni causati dallo stimolo a urinare; difficoltà nello svuotamento della vescica; alterazione dei valori del sangue ai test di laboratorio (per esempio, creatinina o urea).
Raro (riguarda da 1 a 10 utilizzatori su 10'000)
Chetoacidosi diabetica.
Molto raro (riguarda meno di 1 utilizzatore su 10'000)
Fascite necrotizzante del perineo (gangrena di Fournier).
Interrompa l'assunzione di Forxiga e consulti al più presto il suo medico nel caso in cui notasse uno dei seguenti effetti collaterali gravi:
- infezione delle vie urinarie; i segni di una grave infezione delle vie urinarie sono: febbre e/o brividi, bruciore durante la minzione, dolori alla schiena o al fianco;
- reazioni di ipersensibilità (come, ad esempio, gonfiori della pelle o delle mucose, orticaria).
Se osserva effetti collaterali, si rivolga al suo medico o al suo farmacista, soprattutto se si tratta di effetti collaterali non descritti in questo foglietto illustrativo.
Di che altro occorre tener conto?
Il medicamento non dev'essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Indicazione di stoccaggio
Non conservare a temperatura superiore ai 30 °C.
Conservare nella confezione originale.
Conservare fuori dalla portata dei bambini.
Ulteriori indicazioni
Il medico o il farmacista, che sono in possesso di un'informazione professionale dettagliata, possono darle ulteriori informazioni.
Cosa contiene Forxiga?
Principi attivi
1 compressa rivestita con film di Forxiga 5 mg contiene 5 mg di dapagliflozin sotto forma di dapagliflozin propanediolo monoidrato.
1 compressa rivestita con film di Forxiga 10 mg contiene 10 mg di dapagliflozin sotto forma di dapagliflozin propanediolo monoidrato.
Sostanze ausiliarie
Nucleo della compressa
Cellulosa microcristallina (E460i)
Lattosio (25 mg di lattosio nelle compresse rivestite con film da 5 mg. 50 mg di lattosio nelle compresse rivestite con film da 10 mg)
Crospovidone (E1202)
Diossido di silicio (E551)
Magnesio stearato (E470b)
Film di rivestimento
Alcool polivinilico (E1203)
Biossido di titanio (E171)
Macrogol 3350
Talco (E553b)
Ossido di ferro giallo (E172)
Numero dell’omologazione
65176 (Swissmedic)
Dove è ottenibile Forxiga? Quali confezioni sono disponibili?
In farmacia, dietro presentazione della prescrizione medica.
Confezioni da 28 e 98 compresse rivestite con film da 5 mg.
Confezioni da 28 e 98 compresse rivestite con film da 10 mg.
Titolare dell’omologazione
AstraZeneca AG, 6340 Baar
Questo foglietto illustrativo è stato controllato l'ultima volta nel luglio 2020 dall'autorità competente in materia di medicamenti (Swissmedic).
Zusammensetzung
Wirkstoffe
Dapagliflozin als Dapagliflozin-Monohydrat-Propandiol.
Hilfsstoffe
Tablettenkern
Mikrokristalline Cellulose(E460i)
Lactose (25 mg Lactose in 5 mg Filmtabletten. 50 mg Lactose in 10 mg Filmtabletten)
Crospovidon (E1202)
Siliciumdioxid (E551)
Magnesiumstearat (E470b)
Filmüberzug
Poly(vinylalkohol) (E1203)
Titandioxid (E171)
Macrogol 3350
Talkum (E553b)
Gelbes Eisenoxid (E172)
Darreichungsform und Wirkstoffmenge pro Einheit
Filmtabletten mit 5 mg oder 10 mg Dapagliflozin.
Indikationen/Anwendungsmöglichkeiten
Forxiga ist in Ergänzung zu Diät und körperlicher Betätigung bei Erwachsenen (ab 18 Jahren) mit unzureichend kontrolliertem Diabetes mellitus Typ 2 indiziert:
- Als Monotherapie.
- Als Add-on-Kombinationstherapie mit anderen blutzuckersenkenden Arzneimitteln.
- Als initiale Kombinationstherapie mit Metformin.
Für Studienergebnisse zu Kombinationsbehandlungen und Auswirkungen auf kardiovaskuläre Ereignisse siehe Abschnitt «Klinische Wirksamkeit».
Behandlung der Herzinsuffizienz mit reduzierter Auswurffraktion (LVEF ≤40%, NYHA Klasse II-IV) in Ergänzung zu anderen medikamentösen Therapien der Herzinsuffizienz bei adulten Patienten (siehe Abschnitt «Klinische Wirksamkeit»).
Dosierung/Anwendung
Diabetes mellitus
Monotherapie und Add-On Kombinationstherapie
Die empfohlene Anfangsdosis von Forxiga ist 5 mg 1x täglich. Die Dosis kann bei Patienten, die Forxiga 5 mg täglich vertragen und die eine stärkere glykämische Kontrolle benötigen, auf 10 mg täglich erhöht werden. Die Tabletten können unabhängig von den Mahlzeiten zu einer beliebigen Tageszeit eingenommen werden und sind als Ganzes zu schlucken.
Wenn Dapagliflozin in Kombination mit Insulin oder einem insulinotropen Wirkstoff, wie z.B. einem Sulfonylharnstoff, angewendet wird, kann eine niedrigere Dosis des Insulins oder des insulinotropen Wirkstoffs in Erwägung gezogen werden, um das Risiko für eine Hypoglykämie zu senken (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Initiale Kombinationstherapie
Die empfohlene Anfangsdosierung von Dapagliflozin bei Anwendung im Rahmen einer initialen Kombinationstherapie mit Metformin entspricht 5 mg Dapagliflozin einmal täglich.
Herzinsuffizienz
Die empfohlene Dosis von Forxiga zur Behandlung der Herzinsuffizienz beträgt 10 mg einmal täglich. Die Einnahme kann unabhängig von den Mahlzeiten zu einer beliebigen Tageszeit erfolgen.
Forxiga kann zusammen mit anderen Arzneimitteln gegen Herzinsuffizienz angewendet werden.
Patienten mit eingeschränkter Leberfunktion
Eine Dosisanpassung bei Patienten mit leichter oder mässiger Leberfunktionsstörung ist nicht erforderlich. Bei Patienten mit schwerer Leberfunktionsstörung soll mit einer Dosis von 5 mg angefangen werden. Erst wenn diese gut vertragen wird, kann die Dosis auf 10 mg erhöht werden (siehe «Warnhinweise und Vorsichtsmassnahmen» und «Pharmakokinetik»).
Patienten mit eingeschränkter Nierenfunktion
Behandlung von Diabetes mellitus
Bei Patienten, deren geschätzte glomeruläre Filtrationsrate (eGFR) anhaltend bei <45 ml/min/1.73 m2 liegt, sollte Forxiga nicht angewendet werden, da die Wirksamkeit von Dapagliflozin von der Nierenfunktion abhängig ist (siehe «Warnhinweise und Vorsichtsmassnahmen», «Unerwünschte Wirkungen», «Klinische Wirksamkeit» sowie «Pharmakokinetik»).
In der spezifischen Wirksamkeitsstudie an Patienten mit einer eGFR von 45 bis <60 ml/min/1.73 m2 (siehe «Eigenschaften/Wirkungen») wurde ausschliesslich die Dosis von 10 mg pro Tag untersucht. Bei Patienten mit einer eGFR zwischen 45 ml/min/1.73 m2 und <60 ml/min/1.73 m2 sollte eine Therapie mit Dapagliflozin mit der Dosis von 10 mg/Tag eingeleitet werden. Bei Patienten, bei welchen es unter einer Behandlung mit Dapagliflozin zu einer Abnahme der eGFR auf <60 ml/min/1.73 m2 kommt, kann die Therapie hingegen mit der bisherigen Dosierung unter engmaschiger Stoffwechselkontrolle fortgeführt werden.
Bei Patienten mit leichtgradiger Nierenfunktionsstörung ist keine Dosisanpassung erforderlich.
Die 5 mg Dosis wurde nicht in einer Studie geprüft, die spezifisch die Wirksamkeit in Patienten mit einer eGFR von 45-60 ml/min/1.73 m2 untersucht hat.
Behandlung der Herzinsuffizienz
Es sind keine Dosisanpassungen aufgrund der Nierenfunktion erforderlich. Die Anwendung von Forxiga ist bei Patienten mit einer schweren Einschränkung der Nierenfunktion (eGFR <30 ml/min/1.73m2) nicht empfohlen (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Ältere Patienten
Im Allgemeinen wird keine Dosisanpassung aufgrund des Alters empfohlen.
Kinder und Jugendliche
Sicherheit und Wirksamkeit bei Kindern und Jugendlichen sind nicht nachgewiesen. Es liegen keine Daten vor.
Kontraindikationen
Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe dieses Arzneimittels.
Warnhinweise und Vorsichtsmassnahmen
Allgemein
Forxiga sollte nicht bei Patienten mit Diabetes mellitus Typ 1 oder zur Behandlung einer diabetischen Ketoazidose angewendet werden.
Anwendung bei Patienten mit eingeschränkter Leberfunktion
Es liegen begrenzte Erfahrungen aus klinischen Studien zu Patienten mit eingeschränkter Leberfunktion vor. Bei Patienten mit schwerer Leberfunktionsstörung ist die Exposition gegenüber Dapagliflozin erhöht (siehe «Dosierung/Anwendung» und «Pharmakokinetik»). Sicherheit und Wirksamkeit von Dapagliflozin wurden bei Patienten mit schweren Leberfunktionsstörungen nicht untersucht.
Anwendung bei Patienten mit eingeschränkter Nierenfunktion
Es liegen begrenzte Erfahrungen mit Dapagliflozin bei schwerer Nierenfunktionsstörung (eGFR <30 ml/min/1.73 m2) oder terminaler Niereninsuffizienz (ESRD) vor.
Behandlung von Diabetes mellitus
Die Wirksamkeit von Dapagliflozin ist von der Nierenfunktion abhängig. Forxiga sollte daher bei Patienten mit einer eGFR anhaltend <45 ml/min/1.73 m2, nicht zur Behandlung von Diabetes zur Verbesserung der glykämischen Kontrolle angewendet werden.
Überwachung der Nierenfunktion
Die Überwachung der Nierenfunktion wird wie folgt empfohlen:
- vor Therapiebeginn mit Dapagliflozin
- bei Patienten mit normaler Nierenfunktion mindestens einmal jährlich
- bei Patienten mit leichter oder moderater Einschränkung der Nierenfunktion mindestens 2-4mal jährlich (siehe auch «Dosierung/Anwendung», «Unerwünschte Wirkungen», «Eigenschaften/Wirkungen», «Pharmakodynamik» und «Pharmakokinetik»)
- vor der gleichzeitigen Anwendung von Dapagliflozin und einem Arzneimittel, das die Nierenfunktion beeinträchtigen kann; anschliessend periodische Überprüfung.
Falls die eGFR dauerhaft unter 45 ml/min/1.73 m2 abfällt, soll die Therapie mit Dapagliflozin beendet werden.
Anwendung bei Patienten mit Risiko für Hypotonie
Aufgrund des Wirkungsmechanismus induziert Dapagliflozin die osmotische Diurese, was zu der in klinischen Studien beobachteten mässigen Abnahme des Blutdrucks führen kann (siehe «Eigenschaften/Wirkungen»). Diese kann bei Patienten mit sehr hohem Blutzuckerspiegel ausgeprägter sein.
Ketoazidose in Patienten mit Diabetes mellitus
Unter der Behandlung mit Forxiga und anderen SGLT2-Inhibitoren wurden schwerwiegende und teils lebensbedrohliche Fälle diabetischer Ketoazidose (DKA) bei Patienten mit Diabetes mellitus Typ 1 und Typ 2 beobachtet. Forxiga ist nicht für die Behandlung von Patienten mit Diabetes mellitus Typ 1 angezeigt.
Bei Patienten, die unter Behandlung mit Forxiga Symptome wie Übelkeit, Erbrechen, Appetitlosigkeit, Bauchschmerzen, übermässigen Durst, Atembeschwerden, Erschöpfung und Verwirrtheit zeigen, sollten mittels Ketonkörpertest auf das Vorliegen einer Ketoazidose untersucht werden, selbst wenn der Blutzuckerwert bei unter 14 mmol/l (250 mg/dl) liegt. Bei Verdacht auf eine Ketoazidose sollte Forxiga bis zur endgültigen Abklärung abgesetzt werden.
Für Ketoazidose prädisponierende Faktoren umfassen eine niedrige Betazell-Funktionsreserve (z.B. nach vorausgehender Pankreatitis oder Pankreas-Operation), verminderte Kalorienaufnahme, kohlenhydratarme Ernährung, Reduktion der Insulindosis oder steigender Insulinbedarf aufgrund von interkurrenten Erkrankungen (z.B. Infektionen), Operationen oder Alkoholabusus. In solchen Fällen sollte Forxiga mit Vorsicht angewendet werden.
Basierend auf limitierten Daten aus klinischen Studien scheint eine Ketoazidose bei Typ 1-Diabetikern häufiger aufzutreten, wenn diese mit SGLT-2-Inhibitoren behandelt werden. Dapagliflozin soll daher bei Patienten mit Diabetes mellitus Typ 1 nicht eingesetzt werden.
Genitale Infektionen
Bei Patienten mit rezidivierenden mykotischen genitalen Infektionen sollte die Behandlung mit Dapagliflozin überdacht werden, um sicherzustellen, dass der Nutzen der Therapie das Risiko aufwiegt (siehe «Unerwünschte Wirkungen»).
Gleichzeitige Anwendung mit Arzneimitteln, die Hypoglykämien verursachen können
Insulin und insulinotrope Wirkstoffe, etwa Sulfonylharnstoffe, können eine Hypoglykämie induzieren. Daher kann bei gleichzeitiger Verabreichung von Forxiga eine Reduktion der Dosis von Insulin bzw. insulinotropen Wirkstoffen notwendig sein, um das Risiko einer Hypoglykämie zu verringern (siehe «Unerwünschte Wirkungen»).
Fournier-Gangrän
Aus der Arzneimittelüberwachung nach der Markteinführung wurden bei Patienten mit Diabetes Mellitus, die SGLT2-Inhibitoren, einschliesslich Forxiga erhielten, Fälle von nekrotisierender Fasziitis des Perineums (Fournier-Gangrän) berichtet, eine sehr seltene, aber schwerwiegende und potentiell lebensbedrohliche nekrotisierende Infektion, die ein dringendes chirurgisches Eingreifen erfordert. Es waren sowohl Frauen als auch Männer betroffen. Die schwerwiegenden Ausgänge umfassten Krankenhausaufenthalte, mehrere Operationen und Todesfälle.
Patienten, die mit Forxiga behandelt werden und Schmerzen oder Druckempfindlichkeit, Erythem oder Schwellungen im Genital- oder perinealen Bereich sowie Fieber und Unwohlsein aufweisen, sollten auf nekrotisierende Fasziitis untersucht werden. Liegt ein entsprechender Verdacht vor, soll unverzüglich eine Behandlung mit Breitbandantibiotika und gegebenenfalls mit einem chirurgischen Debridement eingeleitet werden. Forxiga sollte abgesetzt und durch eine geeignete Therapiealternative ersetzt werden, dabei sollte der Blutzucker engmaschig überwacht werden.
Hilfsstoffe
Die Filmtabletten enthalten Lactose. Patienten mit einer hereditären Galactose-Intoleranz, Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten dieses Arzneimittel nicht einnehmen.
Interaktionen
Die Metabolisierung von Dapagliflozin erfolgt primär durch die UGT1A9-abhängige Glucuronid-Konjugation.
Die Auswirkungen von Rauchen, Ernährung, pflanzlichen Präparaten und Alkoholkonsum auf die Pharmakokinetik von Dapagliflozin wurden nicht speziell untersucht.
Pharmakokinetische Interaktionen
In-vitro-Studien
In in vitro-Studien hemmte Dapagliflozin weder Cytochrom-P450 (CYP) 1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4 noch induzierte es CYP1A2, CYP2B6 oder CYP3A4. Daher ist nicht zu erwarten, dass Dapagliflozin die metabolische Clearance von gleichzeitig angewendeten Arzneimitteln verändert, die über diese Enzyme metabolisiert werden.
Wirkung anderer Arzneimittel auf Dapagliflozin
In Interaktionsstudien mit gesunden Probanden, in denen hauptsächlich ein Einmalgabe-Design zum Einsatz kam, wurde die Pharmakokinetik von Dapagliflozin nicht verändert durch: Metformin, Pioglitazon, Sitagliptin, Glimepirid, Voglibose, Hydrochlorothiazid, Bumetanid, Valsartan oder Simvastatin.
Rifampicin
Nach gleichzeitiger Verabreichung mit Rifampicin (ein Induktor verschiedener aktiver Transporter und für die Metabolisierung von Arzneimitteln verantwortlicher Enzyme) wurde eine Reduktion der systemischen Exposition gegenüber Dapagliflozin um 22% beobachtet, die aber keine klinisch bedeutsame Wirkung auf die Glucoseausscheidung mit dem Harn über 24 Stunden besass. Daher wird keine Dosisanpassung empfohlen.
Andere Induktoren
Ein klinisch relevanter Effekt mit anderen Induktoren (z.B. Carbamazepin, Phenytoin, Phenobarbital) ist nicht zu erwarten.
Mefenaminsäure
Nach gleichzeitiger Anwendung von Dapagliflozin und Mefenaminsäure (einem UGT1A9-Inhibitor) wurde eine 55%ige Zunahme der systemischen Exposition gegenüber Dapagliflozin beobachtet, jedoch ohne klinisch bedeutsame Auswirkungen auf die Glucoseausscheidung mit dem Harn über 24 Stunden. Es wird keine Dosisanpassung empfohlen.
Wirkung von Dapagliflozin auf andere Arzneimittel
In Interaktionsstudien mit gesunden Probanden, in denen hauptsächlich ein Einmalgabe-Design zum Einsatz kam, veränderte Dapagliflozin nicht die Pharmakokinetik von Metformin, Pioglitazon, Sitagliptin, Glimepirid, Hydrochlorothiazid, Bumetanid, Valsartan, Digoxin (ein P-gp-Substrat) oder Warfarin (S-Warfarin, ein CYP2C9-Substrat) bzw. die anhand der INR gemessenen gerinnungshemmenden Effekte von Warfarin. Die Kombination einer Einmaldosis von Dapagliflozin 20 mg und Simvastatin (ein CYP3A4-Substrat) führte zu einer Zunahme der AUC von Simvastatin um 19% und einer Zunahme der AUC von Simvastatinsäure um 31%. Der Anstieg der Exposition gegenüber Simvastatin und Simvastatinsäure wird als klinisch nicht relevant eingestuft.
Pharmakodynamische Interaktionen
Diuretika
Dapagliflozin kann den diuretischen Effekt von Diuretika verstärken und das Risiko für eine Dehydrierung und eine Hypotonie erhöhen (siehe «Warnhinwiese und Vorsichtsmassnahmen»).
Schwangerschaft/Stillzeit
Schwangerschaft
Es liegen keine Daten zur Anwendung von Dapagliflozin bei Schwangeren vor. Studien an Ratten haben eine Toxizität bezüglich der Nierenausbildung während des Zeitraums gezeigt, der dem zweiten und dritten Schwangerschaftsdrittel beim Menschen entspricht (siehe «Präklinische Daten»). Daher sollte Dapagliflozin während des zweiten und dritten Schwangerschaftsdrittels nicht angewendet werden.
Falls eine Schwangerschaft festgestellt wird, sollte Forxiga abgesetzt werden.
Stillzeit
Es ist nicht bekannt, ob Dapagliflozin und/oder seine Metaboliten beim Menschen in die Muttermilch übergehen. Die vorliegenden Daten zur Pharmakodynamik/Toxikologie im Tierversuch haben ergeben, dass Dapagliflozin/seine Metaboliten in die Milch ausgeschieden werden; zudem haben sich pharmakologisch vermittelte Effekte bei den gesäugten Nachkommen gezeigt (siehe «Präklinische Daten»). Ein Risiko für das Neugeborene/ den Säugling kann nicht ausgeschlossen werden. Forxiga darf deshalb während der Stillzeit nicht angewendet werden.
Fertilität
Die Wirkung von Dapagliflozin auf die Fertilität beim Menschen wurde nicht untersucht.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Forxiga hat keinen oder einen zu vernachlässigenden Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen. Patienten sollten darauf aufmerksam gemacht werden, dass das Risiko für eine Hypoglykämie besteht, wenn Dapagliflozin in Kombination mit einem Sulfonylharnstoff oder Insulin angewendet wird.
Unerwünschte Wirkungen
Zusammenfassung des Sicherheitsprofils
In den klinischen Studien mit Dapagliflozin wurden mehr als 15'000 Patienten mit Typ-2-Diabetes und mehr als 2000 Patienten mit Herzinsuffizienz mit Dapagliflozin behandelt.
Die primäre Bewertung der Sicherheit und Verträglichkeit wurde in einer vorab festgelegten gepoolten Analyse von 13 kurzfristigen (bis zu 24 Wochen) Placebo-kontrollierten Studien durchgeführt, wobei 2'360 Personen mit Dapagliflozin 10 mg und 2'295 mit Placebo behandelt wurden.
Die am häufigsten berichtete unerwünschte Wirkung in allen Studien waren Infektion des Genitalbereichs.
Dapagliflozin 5 mg wurde zusätzlich in einer gepoolten Analyse von 12 Placebo-kontrollierten Kurzzeitstudien evaluiert. Dabei wurden Daten von 1145 Patienten unter Dapagliflozin 5 mg, von 1193 Patienten unter Dapagliflozin 10 mg resp. 1393 Patienten unter Placebo analysiert. Diese Daten stammen aus Monotherapie-Studien und aus Kombinations-Studien mit anderen oralen Antidiabetika.
In der eigens dem kardiovaskulären (CV) Outcome gewidmeten Studie an Patienten mit Typ-2-Diabetes mellitus (DECLARE) erhielten 8'574 Patienten Forxiga 10 mg und 8'569 Patienten Placebo, jeweils über eine mediane Expositionsdauer von 48 Monaten hinweg. Die Forxiga-Exposition belief sich insgesamt auf 30'623 Patientenjahre.
In der Studie mit Dapaglfilozin zum kardiovaskulären Outcome bei Patienten mit Herzinsuffizienz mit reduzierter Auswurffraktion (DAPA-HF) wurden 2368 Patienten mit Dapagliflozin 10 mg und 2368 Patienten über eine mediane Expositionsdauer von 18 Monaten mit Placebo behandelt. Die Patientenpopulation umfasste Patienten mit einer eGFR ≥30 ml/min/1.73 m2 sowie Typ-2-Diabetiker und Nichtdiabetiker.
Das Sicherheitsprofil von Dapagliflozin war insgesamt über die untersuchten Anwendungsgebiete hinweg konsistent. Fälle von diabetischer Ketoazidose wurde ausschliesslich bei Patienten mit Diabetes mellitus beobachtet.
Auflistung der unerwünschten Wirkungen
Die folgenden unerwünschten Wirkungen wurden in den Placebo-kontrollierten klinischen Studien und in Berichten nach der Marktzulassung identifiziert. Keine von ihnen wurde als dosisabhängig befunden. Die unten aufgeführten unerwünschten Wirkungen sind nach Häufigkeit und Systemorganklassen (MedDRA) klassifiziert. Bei den Häufigkeitsangaben werden folgende Kategorien zugrunde gelegt: sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1'000, <1/100), selten (≥1/10'000, <1/1000), sehr selten (<1/10'000).
Infektionen und parasitäre Erkrankungen
Häufig: Vulvovaginitis, Balanitis und verwandte Infektionen des Genitalbereichsa, Harnwegsinfektionenb.
Sehr selten: Fournier-Gangrän (nekrotisierende Fasziitis des Perineums).
Erkrankungen des Blutes und des Lymphsystems
Häufig: erhöhter Hämatokrit.
Stoffwechsel- und Ernährungsstörungen
Sehr häufig: Hypoglykämie (bei Anwendung mit SU oder Insulin).
Häufig*: Volumenmangelc, Dyslipidämie.
Gelegentlich: Durst.
Selten: diabetische Ketoazidosed (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Erkrankungen des Gastrointestinaltrakts
Gelegentlich: Obstipation.
Erkrankungen der Haut und des Unterhautzellgewebes
Gelegentlich: Hyperhidrose.
Unbekannt: Ausschlage.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Rückenschmerzen.
Erkrankungen der Nieren und Harnwege
Häufig: Dysurie, Polyurief.
Gelegentlich: Nykturie, erhöhter Harnstoff im Blut.
Erkrankungen der Geschlechtsorgane und der Brustdrüse
Gelegentlich: Vulvovaginaler Pruritus.
a Vulvovaginitis, Balanitis und verwandte Infektionen des Genitalbereichs schliessen mehrere Standardbegriffe ein, einschliesslich: Vulvovaginale Infektion und Candidose, Balanoposthitis, Balanitis Candida, Penisinfektionen und -Abszesse, bakterielle Vaginitis, Vulvaabszess.
b Harnwegsinfektion schliesst mehrere Standardbegriffe ein, einschliesslich: Urogenitaltraktinfektion, Cystitis, Pyelonephritis, Trigonitis, Urethritis, Prostatitis.
c Volumenmangel schliesst die folgenden Standardbegriffe ein: Dehydrierung, Hypovolämie, Hypotonie.
d Identifiziert in der grossen CV-Outcome Studie bei Patienten mit Typ-2-Diabetes. Häufigkeit basiert auf der jährlichen Rate.
e Ausschlag beinhaltet die folgenden bevorzugten Standardbegriffe: Ausschlag, generalisierter Ausschlag, Ausschlag mit Juckreiz, makulöser Ausschlag, makulo-papulöser Ausschlag, pustulöser Ausschlag, vesikulärer Ausschlag, erythematöser Ausschlag.
f Polyurie schliesst folgende Standardbegriffe ein: Pollakisurie, Polyurie, erhöhte Urinproduktion
Beschreibung ausgewählter Nebenwirkungen
Diabetische Ketoazidose (DKA)
In der DECLARE Studie mit Dapagliflozin bei Patienten mit Typ-2-Diabetes, bei der 8'574 Patienten Dapagliflozin 10 mg und 8'569 Patienten Placebo, mit einer mittleren Expositionszeit von 48 Monaten, erhielten, wurden Ereignisse von DKA bei 27 Patienten in der Dapagliflozin 10 mg-Gruppe und 12 Patienten in der Placebogruppe berichtet. Die Ereignisse traten gleichmässig über den Untersuchungszeitraum verteilt auf. Von den 27 Patienten mit DKA-Ereignissen in der Dapagliflozin-Gruppe hatten 22 zum Zeitpunkt des Ereignisses eine begleitende Insulinbehandlung. Die präzipitierenden Faktoren für DKA waren wie erwartet bei einer Typ-2-Diabetes-Mellitus-Population (siehe Abschnitt «Warnhinweise und Vorsichtsmassnahmen»).
In der DAPA-HF Studie wurde bei 3 Patienten mit Diabetes mellitus Typ 2 in der Dapagliflozin-Gruppe bzw. bei keinem Patienten in der Placebo-Gruppe eine diabetische Ketoazidose berichtet.
Hypoglykämie
Die Häufigkeit von Hypoglykämien hing von der Art der in der jeweiligen Studie angewendeten Hintergrund-Therapie ab.
In Studien mit Dapagliflozin in der Monotherapie, in der Add-on-Therapie mit Metformin oder in der Add-on-Therapie mit Sitagliptin (mit oder ohne Metformin) war die Häufigkeit von leichten Hypoglykämie-Ereignissen bei einer Behandlung bis zu 102 Wochen innerhalb der Behandlungsgruppen, einschliesslich der Placebogruppe, ähnlich (<5%). In allen Studien traten gelegentlich schwere Hypoglykämie-Ereignisse auf und die Inzidenzen waren innerhalb der Gruppen, die mit Dapagliflozin bzw. Placebo behandelt wurden, vergleichbar. Studien zur Add-on-Therapie mit Sulfonylharnstoff und zur Add-on-Therapie mit Insulin wiesen höhere Hypoglykämieraten auf (siehe «Warnhinweise und Vorsichtsmassnahmen»).
In einer Studie zur Add-on-Therapie mit Glimepirid wurde in Woche 24 bzw. Woche 48 über leichte Hypoglykämie-Ereignisse häufiger in der mit Dapagliflozin 10 mg plus Glimepirid behandelten Gruppe (6.0% bzw. 7.9%) und in der mit Dapagliflozin 5 mg plus Glimepirid behandelten Gruppe (5.5% bzw. 8.3%) berichtet als in der mit Placebo plus Glimepirid behandelten Gruppe (2.1% bzw. 2.1%).
In einer Studie zur Add-on-Therapie mit Insulin wurde über schwere Hypoglykämie-Ereignisse in Woche 24 bei 0.5% bzw. in Woche 104 bei 1,0% der Patienten unter Dapagliflozin 10 mg plus Insulin berichtet, in Woche 24 bei 0.5% der Patienten unter Dapagliflozin 5 mg plus Insulin und bei 0.5% in der mit Placebo plus Insulin behandelten Gruppe in der 24. und 104. Woche. Über leichte Hypoglykämie-Ereignisse wurde in Woche 24 bzw. 104 bei 40.3% bzw. 53.1% der Patienten unter Dapagliflozin 10 mg plus Insulin bei 43.4% bzw. 52.8% der Patienten unter Dapagliflozin 5 mg plus Insulin und bei 34.0% bzw. 41.6% der Patienten, die Placebo plus Insulin erhielten.
In der DECLARE Studie mit Dapagliflozin wurde kein erhöhtes Risiko für schwerwiegende Hypoglykämie-Ereignisse bei der Therapie mit Dapagliflozin im Vergleich zu Placebo beobachtet. Schwere Hypoglykämie-Ereignisse wurden bei 58 (0,7%) Patienten in der Dapagliflozin-Gruppe und 83 (1,0%) Patienten in der Placebo-Gruppe gemeldet.
In der DAPA-HF Studie war die Anzahl schwerer Hypoglykämie-Ereignisse in den beiden Behandlungsarmen mit 4 Patienten (0.2%) vergleichbar. Schwere Hypoglykämie-Ereignisse traten nur bei Patienten mit Typ-2-Diabetes auf.
Vulvovaginitis, Balanitis und verwandte Infektionen des Genitalbereichs
Vulvovaginitis, Balanitis und verwandte Infektionen des Genitalbereichs wurden bei 5.5% bzw. 0.6% der Patienten berichtet, die Dapagliflozin 10 mg bzw. Placebo erhielten. Die meisten Infektionen waren leicht bis moderat und führten selten zum Abbruch der Behandlung mit Dapagliflozin, und die Patienten sprachen auf eine Erstbehandlung mit einer Standardtherapie an. Diese Infektionen waren bei Frauen häufiger (8.4% und 1.2% für Dapagliflozin bzw. Placebo). Bei Patienten mit einer entsprechenden Anamnese war eine rezidivierende Infektion wahrscheinlicher.
Im Daten-Pool, bestehend aus den 12 Placebo-kontrollierten Kurzzeitstudien, wurden Vulvovaginitis, Balanitis und verwandte Infektionen des Genitalbereichs bei 0.9% der Patienten unter Placebo, bei 5.7% unter Dapagliflozin 5 mg und bei 4.8% unter Dapagliflozin 10 mg berichtet.
Bei Patienten mit einer entsprechenden Anamnese für genitale Infektionen war eine rezidivierende Infektion wahrscheinlicher.
In der DAPA HF Studie gab es im Dapagliflozin-Arm keine Berichte über schwerwiegende Genitalinfektionen, in der Placebo-Gruppe wurde ein Fall einer schwerwiegenden Genitalinfektion verzeichnet. Die Therapie musste bei sieben Patienten (0.3%) in der Dapagliflozin-Gruppe und bei keinem Patienten in der Placebo-Gruppe wegen eines unerwünschten Ereignisses aufgrund einer Genitalinfektion abgebrochen werden.
Harnwegsinfektionen
Harnwegsinfektionen wurden unter Dapagliflozin 10 mg häufiger als unter Placebo berichtet (4.7% bzw. 3.5%). Die meisten Infektionen waren leicht bis moderat und führten selten zum Abbruch der Behandlung mit Dapagliflozin. Diese Infektionen waren bei Frauen häufiger, und bei Patienten mit einer entsprechenden Anamnese war eine rezidivierende Infektion wahrscheinlicher. Eine Pyelonephritis wurde gelegentlich beobachtet und trat ähnlich häufig auf wie unter der Kontrollbehandlung.
Im Daten-Pool, bestehend aus den 12 Placebo-kontrollierten Kurzzeitstudien, wurden Harnwegsinfektionen bei 3.7% der Patienten unter Placebo, bei 5.7% unter Dapagliflozin 5 mg und bei 4.3% unter Dapagliflozin 10 mg berichtet.
Die Anzahl der Patienten mit einer schwerwiegenden Harnwegsinfektionen war in der DAPA-HF-Studie gering und ausgewogen verteilt: 14 Patienten (0,6%) in der Dapagliflozin- und 17 Patienten (0,7%) in der Placebo-Gruppe. Die Therapie musste bei jeweils 5 Patienten (0,2%) in der Dapagliflozin- und in der Placebo-Gruppe wegen eines unerwünschten Ereignisses aufgrund einer Harnwegsinfektion abgebrochen werden.
Renale unerwünschte Wirkungen
Die Analyse von 13 gepoolten Placebo-kontrollierten Kurzzeitstudien offenbarte einen geringfügigen temporären Anstieg des Serumkreatininins in der Dapagliflozin-Gruppe verglichen mit der Placebo-Gruppe (mittlere Differenz von Ausgangswert in Woche 1 und Woche 24: 0.041 mg/dL versus 0.008 mg/dL und 0.019 mg/dL versus 0.008 mg/dL).
Laboruntersuchungen
Erhöhter Hämatokrit
Im Daten-Pool, bestehend aus den 13 Placebo-kontrollierten Studien, wurde bei Patienten, die Forxiga einnahmen, ein gegenüber dem Ausgangswert erhöhter mittlerer Hämatokrit beobachtet. Dies begann in Woche 1 und dauerte bis Woche 16 an, in welcher der maximale mittlere Unterschied zum Ausganswert beobachtet wurde. In Woche 24 betrug die mittlere Veränderung des Hämatokrits gegenüber dem Ausgangswert 2.30% für Dapagliflozin 10 mg versus -0.33% für Placebo. In Woche 24 wurden Hämatokrit-Werte >55% bei 1.3% der mit Dapagliflozin 10 mg behandelten Patienten und bei 0.4% der mit Placebo behandelten Patienten gemeldet.
Erhöhter Wert des anorganischen Serum-Phosphats
In Woche 24 wurde im Daten-Pool, bestehend aus den 13 Placebo-kontrollierten Studien, bei mit Dapagliflozin behandelten Patienten im Vergleich zu Placebo behandelten Patienten ein gegenüber dem Ausgangswert erhöhter mittlerer Serum-Phosphat-Wert beobachtet (mittlerer Anstieg 0.13 mg/dl versus -0.04 mg/dl). In Woche 24 wiesen unter Forxiga 10 mg 1.7% der Patienten eine Hyperphosphatämie (≥5.6 mg/dl in der Altersgruppe 17 bis 65 Jahre bzw. ≥5.1 mg/dl in der Altersgruppe ≥66 Jahre) auf versus 0.9% unter Placebo.
Auswirkung auf die Lipide
In Woche 24 war im Daten-Pool, bestehend aus den 13 Placebo-kontrollierten Studien, die mittlere prozentuale Veränderung gegenüber dem Ausgangswert für Dapagliflozin 10 mg bzw. Placebo wie folgt: Gesamtcholesterin 2.5% versus 0.0%; HDL Cholesterin 6.0% versus 2.7%; LDL Cholesterin 2.9% versus -1.0%; Triglyzeride -2.7% versus -0.7%.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Dapagliflozin zeigte bei gesunden Probanden nach oralen Einzeldosen von bis zu 500 mg (dem 50-fachen der für den Menschen empfohlenen Höchstdosis) keine Toxizität. Bei diesen Probanden war, abhängig von der Dosis, eine unterschiedlich lange Glukosurie nachweisbar (mindestens über 5 Tage im Fall der 500-mg-Dosis), wobei keine Dehydrierung, Hypotonie oder Elektrolytverschiebungen und kein klinisch bedeutsamer Effekt auf das QTc-Intervall zu beobachten waren. Bei Patienten war die Hypoglykämie-Inzidenz unter Dapagliflozin vergleichbar mit Placebo. In klinischen Studien, in denen bei gesunden Probanden und bei Patienten mit Diabetes mellitus Typ 2 über einen Zeitraum von 2 Wochen Dosen von bis zu 100 mg einmal täglich angewendet wurden (entspricht dem 10-Fachen der für den Menschen empfohlenen Höchstdosis), fiel die Hypoglykämie-Inzidenz geringfügig höher aus als unter Placebo und war nicht dosisabhängig. Die Häufigkeit unerwünschter Ereignisse einschliesslich Dehydrierung oder Hypotonie fiel ähnlich aus wie unter Placebo, und es gab keine klinisch bedeutsamen, dosisabhängigen Veränderungen der Laborwerte, einschliesslich Serumelektrolyten und Biomarkern für die Nierenfunktion.
Behandlung
Im Falle einer Überdosierung sollte in Abhängigkeit vom klinischen Zustand des Patienten eine angemessene supportive Behandlung eingeleitet werden. Die Elimination von Dapagliflozin mittels Hämodialyse wurde nicht untersucht.
Eigenschaften/Wirkungen
ATC-Code
A10BK01
Wirkungsmechanismus
Dapagliflozin ist ein selektiver und reversibler Inhibitor des Natrium/Glucose-Cotransporters 2 (SGLT2), der die glykämische Kontrolle bei Patienten mit Diabetes mellitus verbessert und einen kardio-renalen Nutzen mit sich bringt.
Die von Dapagliflozin bewirkte SGLT2-Hemmung reduziert die Rückresorption von Glucose aus dem glomerulären Filtrat im proximalen renalen Tubulus bei gleichzeitiger Verminderung der Natriumrückresorption. Die Folgen sind Glucoseausscheidung mit dem Urin und osmotische Diurese. Dapagliflozin erhöht somit den Zustrom von Natrium zum distalen Tubulus, was vermutlich zu einer Verstärkung des tubuloglomerulären Feedbacks und zum Absinken des intraglomerulären Drucks führt. Zu den Sekundärwirkungen der SGLT2-Inhibition mit Dapagliflozin gehören ausserdem eine leichte Blutdrucksenkung, Gewichtsabnahme sowie eine Erhöhung des Hämatokrits.
Dapagliflozin verbessert den Nüchtern- sowie den postprandialen Glucosespiegel im Plasma, indem es die renale Glucose-Rückresorption vermindert und so zur Ausscheidung von Glucose im Urin führt. Diese renale Glucoseausscheidung ist bereits nach der ersten Dosis zu beobachten, hält über das 24-stündige Dosierungsintervall an und wird während der gesamten Behandlungsdauer aufrecht erhalten.
Die über diesen Mechanismus renal ausgeschiedene Glucosemenge ist von der Blutglucosekonzentration und der GFR abhängig. Daher besitzt Dapagliflozin bei Probanden mit normalen Glucosespiegeln nur ein geringes Potential für die Induktion einer Hypoglykämie. Die Wirkung von Dapagliflozin ist unabhängig von der Insulinausschüttung und Insulinwirkung.
SGLT2 wird selektiv in der Niere exprimiert. Dapagliflozin besitzt keine Hemmwirkung auf andere Glucosetransporter, die für den Glucosetransport in peripheres Gewebe verantwortlich sind. Dapagliflozin verfügt über eine um den Faktor 1'400 stärkere Selektivität für SGLT2 als für SGLT1, dem wichtigsten für den Glucosetransport verantwortlichen Transporter im Darm.
Pharmakodynamik
Bei gesunden Probanden und bei Patienten mit Diabetes mellitus Typ 2 wurde nach Gabe von Dapagliflozin ein Anstieg der im Urin ausgeschiedenen Glucosemenge verzeichnet. Bei Patienten mit Diabetes mellitus Typ 2 betrug nach 12 Wochen Behandlung mit täglich 10 mg Dapagliflozin die renale Glucoseausscheidung 70 g pro Tag. Die maximale Glucose-Eliminationsrate wurde bei einer Dapagliflozin-Dosis von 20 mg/Tag beobachtet. Bei Patienten mit Diabetes mellitus Typ 2, die bis zu 2 Jahre lang 10 mg Dapagliflozin pro Tag erhielten, zeigte sich eine anhaltende Glukosurie.
Im steady state war die Glucose-Ausscheidung mit dem Harn über 24 Stunden in hohem Mass von der Nierenfunktion abhängig: 85, 52, 18 bzw. 11 g Glucose/Tag wurden von Patienten mit Diabetes mellitus Typ 2 und normaler Nierenfunktion bzw. leichter, moderater oder schwerer Nierenfunktionsstörung ausgeschieden.
Die durch Dapagliflozin induzierte Glukosurie führt bei Patienten mit Diabetes mellitus Typ 2 auch zu osmotischer Diurese und zu einem erhöhten Harnvolumen. Die Erhöhung des Harnvolumens betrug nach einer Behandlung mit täglich 10 mg während 12 Wochen ca. 375 ml/Tag. Der Anstieg des Harnvolumens war mit einer geringfügigen und vorübergehenden Erhöhung der renalen Natriumausscheidung verbunden, die jedoch nicht mit Veränderungen der Natriumkonzentration im Serum einherging.
Auch die renale Harnsäureausscheidung war vorübergehend (über 3-7 Tage) erhöht und ging mit einer anhaltenden Verminderung der Harnsäurespiegel im Serum einher. Nach 24 Wochen lag die Verminderung der Harnsäurespiegel im Serum in einem Bereich von -18.3 bis -48.3 µmol/l (-0.33 bis -0.87 mg/dl).
Klinische Wirksamkeit
Diabetes mellitus
Zur Bewertung der Wirksamkeit und Sicherheit von Forxiga wurden 22 doppelblinde, randomisierte, kontrollierte klinische Studien an insgesamt mehr als 28'000 Patienten mit Typ 2 Diabetes durchgeführt; mehr als 15'000 Patienten wurden in diesen Studien mit Dapagliflozin behandelt. In der Mehrzahl der Studien betrug die Behandlungsdauer 24 Wochen; in anschliessenden offenen Verlängerungen wurden die Patienten über bis zu 156 Wochen behandelt.
In einer gross angelegten CV-Outcome-Studie (DECLARE) wurde die Wirkung von Dapagliflozin auf kardiovaskuläre Ereignisse bei Patienten mit Typ-2-Diabetes mellitus mit oder ohne manifeste kardiovaskuläre Erkrankung untersucht.
Glykämische Kontrolle
Monotherapie
Um die Sicherheit und Wirksamkeit einer Monotherapie mit Dapagliflozin bei Patienten mit Diabetes mellitus Typ 2 zu bewerten, wurde eine doppelblinde, Placebo-kontrollierte Studie über eine Dauer von 24 Wochen (mit einer zusätzlichen Verlängerungsperiode) durchgeführt Die einmal tägliche Behandlung mit Dapagliflozin führte im Vergleich zu Placebo zu einer statistisch signifikanten Reduktion des HbA1c-Wertes (siehe Tabelle 1). In der Verlängerungsperiode wurden die HbA1c-Reduktion bis Woche 102 aufrechterhalten (-0.63%, -0.77% bzw. -0.18% adjustierte mittlere Veränderung gegenüber dem Ausgangswert für Dapagliflozin 10 mg, Dapagliflozin 5 mg bzw. Placebo).
Tabelle 1: Ergebnisse einer Placebo-kontrollierten Studie mit Dapagliflozin als Monotherapie in Woche 24 (LOCFa)
Monotherapie | |||
Dapagliflozin | Dapagliflozin | Placebo | |
Nb | 70 | 64 | 75 |
HbA1c (%) | |||
Mittlerer Ausgangswert | 8.01 | 7.83 | 7.79 |
Veränderung zum Ausgangswertc | -0.89 | -0.77 | -0.23 |
Differenz zu Placeboc (95% KI) | -0.66* (-0.96; -0.36) | -0.54* (-0.84; -0.24) | |
Körpergewicht (kg) | |||
Mittlerer Ausgangswert | 94.13 | 87.17 | 88.77 |
Veränderung zum Ausgangswertc | -3.16 | -2.83 | -2.19 |
Differenz zu Placeboc (95% KI) | -0.97 (-2.20; 0.25) | -0.65 (-1.90; 0.61) | |
a LOCF (last observation carried forward): letzter vorliegender Wert für jeden Patienten (bei Patienten mit Rescue-Therapie vor der Rescue-Therapie)
b Alle randomisierten Personen, die während der doppelblinden Kurzzeitphase mindestens eine Dosis der doppelblinden Studienmedikation einnahmen
c Least-Squares-Mittelwert, adjustiert nach Ausgangswert
* p-Wert <0.0001 versus Placebo
Kombinationstherapie
In einer 52-wöchigen, Verum-kontrollierten Nicht-Unterlegenheitsstudie (mit einer 52-wöchigen Verlängerungsperiode) wurde Dapagliflozin bei Personen mit unzureichender glykämischer Kontrolle (HbA1c >6.5% und ≤10%) als Add-on-Therapie zu Metformin im Vergleich zu einem Sulfonylharnstoff (Glipizid) als Add-on-Therapie zu Metformin bewertet. Die Ergebnisse zeigten im Vergleich zu Glipizid eine ähnliche mittlere Reduktion des HbA1c-Wertes gegenüber dem Ausgangswert bis Woche 52 und belegen so die Nicht-Unterlegenheit (siehe Tabelle 2). In Woche 104 betrug die adjustierte mittlere Veränderung des HbA1c-Wertes gegenüber dem Ausgangswert -0.32% für Dapagliflozin und -0.14% für Glipizid. Innerhalb der 52 bzw. 104 Wochen trat mindestens ein hypoglykämisches Ereignis bei einem signifikant kleineren Anteil an Patienten in der mit Dapagliflozin behandelten Gruppe (3.5% bzw. 4.3%) auf im Vergleich zur Gruppe, die mit Glipizid behandelt wurde (40.8% bzw. 47.0%). In Woche 104 waren in der Dapagliflozin-Gruppe noch 56.2%, in der Glipizid-Gruppe noch 50.0% der Patienten in der Studie verblieben.
Tabelle 2: Ergebnisse einer aktiv kontrollierten Studie zum Vergleich von Dapagliflozin und Glipizid als Add-on-Therapie mit Metformin in Woche 52 (LOCFa)
Parameter | Dapagliflozin | Glipizid |
|---|---|---|
| Nb | 400 | 401 |
HbA1c (%) | ||
| Mittlerer Ausgangswert | 7.69 | 7.74 |
| Veränderung zum Ausgangswertc | -0.52 | -0.52 |
| Differenz zu Glipizid + Metforminc (95% KI) | 0.00d (-0.11; 0.11) | |
Körpergewicht (kg) | ||
| Mittlerer Ausgangswert | 88.44 | 87.60 |
| Veränderung zum Ausgangswertc | -3.22 | 1.44 |
| Differenz zu Glipizid + Metforminc (95% KI) | -4.65* (-5.14; -4.17) | |
a LOCF: letzter vorliegender Wert für jeden Patienten
b Randomisierte und behandelte Personen mit Ausgangswert und mindestens 1 Wirksamkeitsmessung nach Ausgangswert
c Least-Squares-Mittelwert, adjustiert nach Ausgangswert
d nicht unterlegen gegenüber Glipizid + Metformin
* p-Wert <0.0001
Dapagliflozin als Add-on zu entweder Metformin, Glimepirid, Sitagliptin (mit oder ohne Metformin) oder Insulin führte zu statistisch signifikanten Reduktionen des HbA1c-Wertes in Woche 24 (p<0.0001; siehe Tabellen 3 bis 6) verglichen mit Patienten, die Placebo erhielten.
Die in Woche 24 beobachtete Reduktion des HbA1c-Wertes blieb in den Add-on-Kombinationsstudien (Glimepirid und Insulin) gemäss den 48-Wochen-Daten (Glimepirid) und den bis zu 104-Wochen-Daten (Insulin) erhalten. In der Add-on-Therapie mit Sitagliptin (mit oder ohne Metformin) betrug die adjustierte mittlere Veränderung gegenüber dem Ausgangswert in Woche 48 für Dapagliflozin 10 mg und Placebo -0.30% bzw. 0.38%. Die Reduktion des HbA1c-Wertes im Rahmen der Studie zur Add-on-Therapie mit Metformin blieb bis Woche 102 erhalten (adjustierte mittlere Veränderung gegenüber dem Ausgangswert von -0.58%, -0.78% und 0.02% für 5 mg, 10 mg bzw. Placebo).
In der Studie mit Insulin (mit oder ohne zusätzlichem oral blutzuckersenkendem Arzneimittel) betrug in Woche 104 die adjustierte mittlere Veränderung des HbA1c-Wertes gegenüber dem Ausgangswert ‑0.71% für Dapagliflozin 5 mg, -0.71% für Dapagliflozin 10 mg bzw. -0.06% für Placebo. Bei Personen, die mit Dapagliflozin 5 mg oder 10 mg behandelt wurden, blieb die Insulin-Dosis mit einer mittleren Dosis im Bereich von 75 - 79 IU/Tag in Woche 48 und 104 im Vergleich zum Ausgangswert stabil. In der Placebo-Gruppe betrug die mittlere Erhöhung gegenüber dem Ausgangswert 10.5 IU/Tag in Woche 48 bzw. 18.3 IU/Tag in Woche 104 (mittlere Dosis von 84 IU/Tag und 92 IU/Tag). In Woche 104 waren in der Dapagliflozin-Gruppe noch 72.4%, in der Glipizid-Gruppe noch 54.8% der Patienten in der Studie verblieben.
Tabelle 3: Ergebnisse von 24-wöchigen (LOCFa), Placebo-kontrollierten Studien mit Dapagliflozin als Add-on-Kombination mit Metformin
Add-on-Kombination | |||
Metformin1 | |||
Dapagliflozin | Dapagliflozin | Placebo | |
Nb | 135 | 137 | 137 |
HbA1c (%) | |||
Mittlerer Ausgangswert | 7.92 | 8.17 | 8.11 |
Veränderung zum Ausgangswertc | -0.84 | -0.70 | -0.30 |
Differenz zu Placeboc (95% KI) | -0.54* (-0.74; -0.34) | -0.41* (-0.61; -0.21) | |
Personen (%), die einen HbA1c <7% erreichen: Adjustiert nach Ausgangswert | 40.6** | 37.5** | 25.9 |
Körpergewicht (kg) | |||
Mittlerer Ausgangswert | 86.28 | 84.73 | 87.74 |
Veränderung zum Ausgangswertc | -2.86 | -3.04 | -0.89 |
Differenz zu Placeboc (95% KI) | -1.97* (-2.63; -1.31) | -2.16* (-2.81; -1.50) | |
1 Metformin ≥1500 mg/Tag
a LOCF: letzter vorliegender Wert für jeden Patienten (bei Patienten mit Rescue-Therapie vor der Rescue-Therapie)
b Alle randomisierten Personen, die während der doppelblinden Kurzzeitphase mindestens eine Dosis der doppelblinden Studienmedikation einnahmen
c Least-Squares-Mittelwert, adjustiert nach Ausgangswert
* p-Wert <0.0001 versus Placebo + orales blutzuckersenkendes Arzneimittel
** p-Wert <0.05 versus Placebo + orales blutzuckersenkendes Arzneimittel
Tabelle 4: Ergebnisse von 24-wöchigen (LOCFa), Placebo-kontrollierten Studien mit Dapagliflozin als Add-on-Kombination mit Glimepirid
Add-on-Kombination | |||
Sulfonylharnstoff (Glimepirid1) | |||
Dapagliflozin 10 mg | Dapagliflozin 5 mg | Placebo | |
Nb | 151 | 142 | 145 |
HbA1c (%) | |||
Mittlerer Ausgangswert | 8.07 | 8.12 | 8.15 |
Veränderung zum Ausgangswertc | -0.82 | -0.63 | -0.13 |
Differenz zu Placeboc (95% KI) | -0.68* (-0.86; -0.51) | -0.49* (-0.67; -0.32) | |
Personen (%), die einen HbA1c <7% erreichen: Adjustiert nach Ausgangswert | 31.7* | 30.3* | 13.0 |
Körpergewicht (kg) | |||
Mittlerer Ausgangswert | 80.56 | 81.00 | 80.94 |
Veränderung zum Ausgangswertc | -2.26 | -1.56 | -0.72 |
Differenz zu Placeboc (95% KI) | -1.54* (-2.17; -0.92) | -0.84* (-1.47; -0.21) | |
1 Glimepirid 4 mg/Tag
a LOCF: letzter vorliegender Wert für jeden Patienten (bei Patienten mit Rescue-Therapie vor der Rescue-Therapie)
b Alle randomisierten Personen, die während der doppelblinden Kurzzeitphase mindestens eine Dosis der doppelblinden Studienmedikation einnahmen
c Least-Squares-Mittelwert, adjustiert nach Ausgangswert
* p-Wert <0.0001 versus Placebo + orales blutzuckersenkendes Arzneimittel
Tabelle 5: Ergebnisse von 24-wöchigen (LOCFa), Placebo-kontrollierten Studien mit Dapagliflozin als Add-on-Kombination mit Sitagliptin (mit oder ohne Metformin)
Add-on-Kombination | ||
DPP-4-Inhibitor (Sitagliptin2) ± Metformin1 | ||
Dapagliflozin 10 mg | Placebo | |
Nb | 223 | 224 |
HbA1c (%) | ||
Mittlerer Ausgangswert | 7.90 | 7.97 |
Veränderung zum Ausgangswertc | -0.45 | 0.04 |
Differenz zu Placeboc (95% KI) | -0.48* (-0.62; -0.34) | |
Körpergewicht (kg) | ||
Mittlerer Ausgangswert | 91.02 | 89.23 |
Veränderung zum Ausgangswertc | -2.14 | -0.26 |
Differenz zu Placeboc (95% KI) | -1.89* (-2.37; -1.40) | |
1 Metformin ≥1500 mg/Tag
2 Sitagliptin 100 mg/Tag
a LOCF: letzter vorliegender Wert für jeden Patienten (bei Patienten mit Rescue-Therapie vor der Rescue-Therapie)
b Alle randomisierten Personen, die während der doppelblinden Kurzzeitphase mindestens eine Dosis der doppelblinden Studienmedikation einnahmen
c Least-Squares-Mittelwert, adjustiert nach Ausgangswert
* p-Wert <0.0001 versus Placebo + orales blutzuckersenkendes Arzneimittel
Tabelle 6: Ergebnisse einer Placebo-kontrollierten Studie mit Dapagliflozin in Kombination mit Insulin (allein oder mit oralen blutzuckersenkenden Arzneimitteln) in Woche 24 (LOCFa)
Parameter | Dapagliflozin 10 mg | Dapagliflozin 5 mg | Placebo |
Nb | 194 | 211 | 193 |
HbA1c (%) | |||
Mittlerer Ausgangswert | 8.58 | 8.61 | 8.46 |
Veränderung zum Ausgangswertc | -0.90 | -0.82 | -0.30 |
Differenz zu Placeboc (95% KI) | -0.60* (-0.74; -0.45) | -0.52* (-0.66; -0.38) | |
Körpergewicht (kg) | |||
Mittlerer Ausgangswert | 94.63 | 93.20 | 94.21 |
Veränderung zum Ausgangswertc | -1.67 | -0.98 | 0.02 |
Differenz zu Placeboc (95% KI) | -1.68* (-2.19; -1.18) | -1.00* (-1.50; -0.50) | |
Mittlere tägliche Insulin-Dosis (IU)1 | |||
Mittlerer Ausgangswert | 77.96 | 77.10 | 73.96 |
Veränderung zum Ausgangswertc | -1.16 | -0.61 | 5.08 |
Differenz zu Placeboc (95% KI) | -6.23* (-8.84; -3.63) | -5.69* (-8.25; -3.13) | |
a LOCF: letzter vorliegender Wert für jeden Patienten (vor oder am Tag der ersten Insulin-Auftitration, falls erforderlich)
b Alle randomisierten Personen, die während der doppelblinden Kurzzeitphase mindestens eine Dosis der doppelblinden Studienmedikation einnahmen
c Least-Squares-Mittelwert, adjustiert nach Ausgangswert und Vorhandensein eines oralen blutzuckersenkenden Arzneimittels
* p-Wert <0.0001 versus Placebo + Insulin ± orales blutzuckersenkendes Arzneimittel
1 Auftitration des Insulin-Regimes (einschliesslich kurz wirksamen, intermediären und basalen Insulins) war nur erlaubt, wenn Personen vordefinierte FPG-Kriterien erfüllten.
2 Fünfzig Prozent der Personen erhielten zum Ausgangszeitpunkt Insulin als Monotherapie; 50% erhielten 1 oder 2 orale blutzuckersenkende Arzneimittel zusätzlich zu Insulin: Von dieser letztgenannten Gruppe erhielten 80% Metformin allein, 12% erhielten eine Therapie mit Metformin plus Sulfonylharnstoff und die Übrigen erhielten andere orale blutzuckersenkende Arzneimittel.
Kombination von Dapagliflozin und GLP-1-Rezeptoragonist (GLP-1-RA)
Eine 28-wöchige, 3-armige, randomisierte (1:1:1), doppelblinde Studie untersuchte die glukosesenkende Wirkung der Kombination von Dapagliflozin (10 mg einmal täglich) und einem GLP-1-RA (Exenatid Retard 2 mg einmal wöchentlich) und der Monotherapien mit dem GLP-1-RA bzw. Dapagliflozin. Die Studie schloss 694 erwachsene Patienten mit Typ-2-Diabetes mellitus und unzureichender Blutzuckerkontrolle (HbA1c ≥8.0 und ≤12.0%) unter vorheriger Metformin-Monotherapie (≥1.500 mg/Tag; während der Behandlungsphase unverändert fortgesetzt) ein. Primärer Endpunkt war die Änderung des HbA1c von Studienbeginn bis Woche 28. Die Kombination aus Dapagliflozin und GLP-1-RA war den beiden Monotherapien hinsichtlich der Senkung des HbA1c überlegen (siehe Tabelle 7).
Tabelle 7: Glukosesenkende Wirkung von Dapagliflozin in Kombination mit Exenatid Retard, Dapagliflozin und Exenatid Retard in Metformin-behandelten Patienten (28-Wochen Behandlungsphase)
Parameter | Dapagliflozin 10 mg QD + | Dapagliflozin 10 mg QD + | Exenatid retard 2 mg QW + |
Nc | 228 | 230 | 227 |
HbA1c (%) | |||
Mittlerer Ausgangswert | 9.29 | 9.25 | 9.26 |
Veränderung zum Ausgangswerta | -1.98 | -1.39 | -1.60 |
Differenz zu Placebo (95% KI) | -0.59* (-0.84, -0.34) | ||
Differenz zu Exenatid retard QW (95% KI) | -0.38§ (-0.63, -0.13) | ||
Anteil Patienten mit Erreichung von HbA1c <7.0%b | 44.7% | 19.1% | 26.9% |
QD = einmal täglich, QW = einmal wöchentlich, N = Anzahl Patienten in der Behandlungsgruppe, KI = Konfidenzintervall.
a Modellierung der adjustierten Kleinste-Quadrate-Mittelwerte (KQ-Mittel) und Behandlungsgruppendifferenz(en) für die Veränderung gegenüber dem Ausgangswert in Woche 28 erfolgte mit einem gemischten Modell mit wiederholten Messungen (MMRM; mixed model with repeated measures) unter Berücksichtigung von Behandlung, Region, Stratum HbA1c-Ausgangswert (<9.0% oder ≥9.0%), Woche und Interaktion Behandlung–Woche als feste Faktoren und dem Ausgangswert als Kovariate.
b Kategorien abgeleitet aus kontinuierlichen Messungen. Bei allen Patienten mit fehlenden Endpunktdaten Imputation als Nonresponder. Behandlungsvergleich durch Cochran-Mantel-Haenszel (CMH)-Test, stratifiziert nach HbA1c-Ausgangswert (<9.0% oder ≥9.0%). P-Werte gemäss allgemeiner Assoziationsstatistik.
c Patienten, die mindestens einmal die Studienmedikation erhielten und für die mindestens ein post-baseline HbA1c-Messwert vorliegt.
* p <0.001.
§ p <0.01.
Alle p-Werte sind adjustierte p-Werte für Multiplizität.
Von den Analysen ausgeschlossen waren Messungen nach Rescue-Therapie und nach vorzeitiger Absetzung der Studienmedikation.
Initiale Kombinationstherapie mit Metformin
In zwei kontrollierten 3-armigen 24-wöchigen Studien wurde die Wirksamkeit und Sicherheit einer Behandlung mit Metformin XR, Dapagliflozin, Metformin XR plus Dapagliflozin in insgesamt 1'236 Therapie-naiven Patienten mit unzureichend kontrolliertem Typ-2-Diabetes (HbA1c ≥7.5% und ≤12%) untersucht. Die Metformin XR Dosis war 2000 mg in beiden Studien. Die Dapagliflozin Dosis war 5 mg bzw. 10 mg in der jeweiligen Studie. In allen 3 Behandlungsarmen beider Studien kam es zu einer signifikanten Abnahme des HbA1c; dabei war die Kombinationstherapie (Metformin XR plus Dapagliflozin) in beiden Studien den Monotherapien mit Metformin XR oder Dapagliflozin überlegen (Tabelle 8).
Tabelle 8: Ergebnisse zweier kontrollierter Studien mit Dapagliflozin im Rahmen einer initialen Kombinationstherapie mit Metformin XR (24-Wochen Behandlungsphase)
Parameter | Dapagliflozin + Metformin XR | Dapagliflozin + Placebo | Metformin XR + Placebo | |||
Dapagliflozin Dosierung in Studie | 5 mg | 10 mg | 5 mg | 10 mg | 5 mg | 10 mg |
N† | 194 | 211 | 203 | 219 | 201 | 208 |
HbA1c (%) | ||||||
Mittlerer Ausgangswert | 9.21 | 9.10 | 9.14 | 9.03 | 9.14 | 9.03 |
Veränderung ggü. Ausgangswert (adjustiertes Mittel‡) | -2.05 | -1.98 | -1.19 | -1.45 | -1.35 | -1.44 |
Unterschied ggü. Dapagliflozin (adjustiertes Mittel‡) (95% KI) | -0.86§ (-1.11,-0.62) | -0.53§ (-0.74, -0.32) | ||||
Unterschied ggü. Metformin XR (adjustiertes Mittel‡) (95% KI) | -0.70§ (-0.94,-0.45) | -0.54§ (-0.75, -0.33) | -0.01¶ (-0.22, 0.20) | |||
Anteil Patienten, die einen HbA1c <7,0% erreichen, adjustiert für den Ausgangswert | 52.4% | 46.6%# | 22.5% | 31.7% | 34.6% | 35.2% |
HbA1c-Veränderung ggü. Ausgangswert bei Patienten mit einem HbA1c-Ausgangswert ≥9% (adjustiertes Mittel‡) | -3.01 | -2.59# | -1.67 | -2.14 | -1.82 | -2.05 |
Unterschied ggü. Dapagliflozin (adjustiertes Mittel‡) (95% KI) | -1.34§ (-1.72, -0.96) | -0.45# (-0.81, -0.09) | ||||
Unterschied ggü. Metformin XR (adjustiertes Mittel‡) (95% KI) | -1.19§ (-1.57, -0.82) | -0.53 (-0.89, -0.18) | ||||
* LOCF: last observation carried forward: Der letzte (vor der eventuellen Durchführung einer Rescue-Therapie) vorliegende Wert für jeden Patienten wurde für die Endauswertung herangezogen.
† Alle randomisierten Patienten, die während der doppelblinden Kurzzeit-Phase mindestens eine Dosis der doppelblinden Studienmedikation eingenommen haben.
‡ Für den Ausgangswert adjustierter Kleinstquadratmittelwert.
§ p-Wert <0.0001.
¶ Nichtunterlegen versus Metformin XR.
# p-Wert <0.05.
Nüchtern-Plasma-Glucose (fasting plasma glucose, FPG)
Die Behandlung mit Dapagliflozin 5 mg oder 10 mg als Monotherapie oder als Add-on-Therapie mit entweder Metformin, Glimepirid oder Sitagliptin (mit oder ohne Metformin) führte zu einer statistisch signifikanten Reduktion der Nüchtern-Plasma-Glucose (-1.90 bis -1.18 mmol/l) verglichen mit Placebo (‑0.33 bis 0.21 mmol/l). Dieser Effekt wurde in Woche 1 der Behandlung beobachtet und hielt in Studien, die bis Woche 102 fortgeführt wurden, an.
Postprandiale Glucose
Die Behandlung mit Dapagliflozin 10 mg als Add-on-Therapie mit Glimepirid führte bis Woche 24 zu einer statistisch signifikanten Reduktion der postprandialen Glucose (2 Stunden nach der Mahlzeit), die bis Woche 48 anhielt.
Die Behandlung mit Dapagliflozin 10 mg als Add-on-Therapie mit Sitagliptin (mit oder ohne Metformin) führte bis Woche 24 zu einer Reduktion der postprandialen Glucose (2 Stunden nach der Mahlzeit), die bis Woche 48 anhielt.
Blutdruck
Die Behandlung mit Dapagliflozin 10 mg führte gemäss Analyse der gepoolten Daten von 13 Placebo-kontrollierten Studien in Woche 24 zu einer Veränderung des systolischen Blutdrucks gegenüber dem Ausgangswert um -3.7 mmHg und des diastolischen Blutdrucks um -1.8 mmHg versus -0.5 mmHg beim systolischen und -0.5 mmHg beim diastolischen Blutdruck in der Placebo-Gruppe. Dieser Effekt blieb bis Woche 104 erhalten. Im Daten-Pool, bestehend aus den 12 Placebo-kontrollierten Kurzzeitstudien, führte die Behandlung mit Dapagliflozin 5 mg zu einer Veränderung des systolischen Blutdrucks gegenüber dem Ausgangswert um -3.5 mmHg und des diastolischen Blutdrucks um -2.1 mmHg.
Patienten mit Nierenfunktionsstörung
Moderate Nierenfunktionsstörung (eGFR ≥45 bis <60 ml/min/1.73 m2)
In einer doppelblinden, randomisierten, placebokontrollierten Studie wurde die Wirksamkeit von Dapagliflozin an insgesamt 321 erwachsenen Patienten mit Diabetes mellitus Typ 2 und einer eGFR von ≥45 bis <60 ml/min/1.73 m2 (d.h. mit moderater Nierenfunktionsstörung entsprechend CKD 3A) sowie einer unter der aktuellen Behandlung unzureichenden Blutzuckereinstellung untersucht. Die Patienten wurden entweder mit 10 mg Dapagliflozin (n=159) oder mit Placebo (n=161) behandelt. Das HbA1c der eingeschlossenen Patienten lag bei Studienbeginn bei durchschnittlich 8.2%. Im Primärendpunkt, der Veränderung des HbA1c gegenüber Baseline, fand sich in Woche 24 für 10 mg Dapagliflozin eine statistisch signifikante Überlegenheit gegenüber Placebo. Die mittlere Veränderung des HbA1c gegenüber dem Ausgangswert betrug unter Dapagliflozin -0.37%, unter Placebo -0.03%, entsprechend einer Therapiedifferenz von -0.34% (95% KI -0.53, -0.15).
Das Sicherheitsprofil von Dapagliflozin entsprach jenem früherer Studien in der Gesamtpopulation von Patienten mit Diabetes mellitus Typ 2. In der Dapagliflozin-Gruppe nahm die mittlere eGFR zu Beginn der Behandlung ab (Dapagliflozin: -3.39 ml/min/1.73 m2, Placebo: -0.90 ml/min/1.73 m2) und blieb dann während der anschliessenden 24-wöchigen Behandlungsphase stabil. Drei Wochen nach Absetzen von Dapagliflozin fanden sich keine relevanten Unterschiede mehr in der eGFR zwischen Verum- und Placebo-Gruppe.
In eine frühere Studie an insgesamt n=252 Diabetikern waren zusätzlich auch Patienten mit einer eGFR zwischen 30 und <45 ml/min/1.73 m2 eingeschlossen (durchschnittliche eGFR in dieser Studie 45 ml/min/1.73 m2). In dieser Studie konnte für Dapagliflozin weder für die 5 mg-Dosis noch für die 10 mg-Dosis eine Wirksamkeit gezeigt werden (siehe «Dosierung / Anwendung»).
Patienten mit einem HbA1c-Ausgangswert ≥9%
Bei Patienten mit einem HbA1c-Ausgangswert ≥9.0% führte die Behandlung mit Dapagliflozin 10 mg zu einer statistisch signifikanten Reduktion des HbA1c-Wertes in Woche 24 sowohl in der Monotherapie (adjustierte mittlere Veränderung zum Ausgangswert: -2.04% und 0.19% für Dapagliflozin 10 mg bzw. Placebo) als auch in der Add-on-Behandlung mit Metformin (adjustierte mittlere Veränderung zum Ausgangswert: -1.32% und -0.53% für Dapagliflozin bzw. Placebo).
Kardiovaskuläre Outcome Studie (DECLARE)
Die Wirkung einer Dapagliflozin-Behandlung auf kardiovaskuläre Ereignisse wurden über einen medianen Beobachtungszeitraum von 4.2 Jahren in einer internationalen, multizentrischen, doppelblinden kardiovaskulären Outcome-Studie (DECLARE) untersucht, in welcher die Patienten mit Typ-2-Diabetes (mittlere Erkrankungsdauer 11.9 Jahre) zusätzlich zur vorbestehenden Therapie randomisiert mit Dapagliflozin (n=8582) oder Placebo (n=8578) behandelt wurden. Die Pharmakotherapie des Diabetes und sonstiger Komorbiditäten konnte während der Studie gemäss «Standard of Care (SoC)» angepasst werden.
Der mittlere HbA1c bzw. BMI zu Studienbeginn betrug 8.3% bzw. 32.1 kg/m2. Die Studienpopulation mit einem Durchschnittsalter von 63.9 Jahren bestand zu 59.4% aus Patienten ohne CV Vorerkrankung mit folgenden Risikofaktoren: Alter ≥55 (Männer) bzw. ≥60 Jahre (Frauen) sowie Dyslipidämie, Hypertonie oder Rauchen. Die restlichen 40.6% der Patienten wiesen bereits zu Studienbeginn eine manifeste CV Erkrankung auf. Bei 10.0% der Patienten war eine Herzinsuffizienz anamnestisch belegt. Die mittlere eGFR betrug 85.2 ml/min/1.73 m2 (nur 7.4% der Patienten hatten eine eGFR <60ml/min/1.73 m2), während 30.3% der Patienten eine Albuminurie aufwiesen.
Zu Studienbeginn erhielten 82.0% der Patienten Metformin zur Senkung des Blutzuckers; weitere antihyperglykämische Behandlungen schlossen Insulin (40.9%), Sulfonylharnstoffe (42.7%), DPP4-Inhibitoren (16.8%) und GLP-1-Agonisten (4.4%) ein. Zur Behandlung CV Komorbiditäten erhielten die Patienten ACE-Inhibitoren bzw. Angiotensin-II-Rezeptor-Blockern (81.3%), Statinen (75.0%), Thrombozytenaggregationshemmern (61.1%), Acetylsalicylsäure (55,5%), Betablockern (52.6%), Calciumkanalblockern (34.9%), Thiaziddiuretika (22.0%) und Schleifendiuretika (10.5%).
Die Ergebnisse der primären Analyse sind in Tabelle 9 zusammengefasst.
Endpunkt | Dapagliflozin (n=8582) | Placebo (n=8578) | HR [95% CI] | ||
Patienten mit Ereignis | Ereignisrate (per 1000 Patientenjahre) | Patienten mit | Ereignisrate (per 1000 Patientenjahre) | ||
MACE | 756 (8.8) | 22.6 | 803 (9.4) | 24.2 | 0.93 [0.84; 1.03] |
CV Tod | 245 (2.9) | 7.0 | 249 (2.9) | 7.1 | 0.98 [0.82; 1.17] |
Myokardinfarkt | 393 (4.6) | 11.7 | 441 (5.1) | 13.2 | 0.89 [0.77; 1.01] |
Hirnschlag | 235 (2.7) | 6.9 | 231 (2.7) | 6.8 | 1.01 [0.84; 1.21] |
Herzinsuffizienz- Composite | 417 (4.9) | 12.2 | 496 (5.8) | 14.7 | 0.83 [0.73; 0.95] |
Hospitalisierung aufgrund von Herzinsuffizienz | 212 (2.5) | 6.2 | 286 (3.3) | 8.5 | 0.73 [0.61; 0.88] |
CV Tod | 245 (2.9) | 7.0 | 249 (2.9) | 7.1 | 0.98 [0.82; 1.17] |
Herzinsuffizienz
Dapagliflozin And Prevention of Adverse outcomes in Heart Failure (DAPA-HF) war eine internationale, multizentrische, randomisierte, doppelblinde placebokontrollierte Studie an insgesamt 4744 Patienten mit Herzinsuffizienz (New York Heart Association [NYHA] Funktionsklasse II-IV) mit reduzierter Auswurffraktion (linksventrikuläre Auswurffraktion [LVEF] ≤40%) zur Untersuchung der Wirkung von Dapagliflozin als Zusatz zur Standard-Hintergrundtherapie auf CV-bedingte Mortalität und Verschlechterung der Herzinsuffizienz (Hospitalisierung wegen Herzinsuffizienz und dringender Arztbesuch wegen Herzinsuffizienz).
Patienten, die mit einer dem Therapiestandard entsprechenden Herzinsuffizienztherapie behandelt wurden (Tabelle 10), wurden Dapagliflozin 10 mg (n=2373) oder Placebo (n=2371) zugeteilt.
Tabelle 10: Begleitende Herzinsuffizienztherapie bei Studienbeginn
Begleitende Herzinsuffizienztherapie | Prozent der Patienten bei Studienbeginn (%) |
ACE Inhibitor oder Angiotensin-Rezeptorblocker oder Angiotensin-Rezeptor-Neprilysin-Inhibitor (ARNI) | 94% |
ARNI | 11% |
Betablocker | 96% |
Mineralocorticoid-Rezeptor-Antagonisten (MRA) | 71% |
Diuretikum | 93% |
Implantierbares Gerät | 26% |
Das Hauptziel der Studie bestand in der Beantwortung der Frage, ob Dapagliflozin kardiovaskulär bedingte Todesfälle und die Verschlechterung der Herzinsuffizienz verhindert und es Herzinsuffizienzsymptome verbessert.
Der Beobachtungszeitraum erstreckte sich über median 18 Monate. Das Patientenkollektiv war im Mittel 66 Jahre alt und zu 77% männlich; 70% der Patienten waren Weisse, 5% Schwarze oder Afroamerikaner und 24% Asiaten.
Zu Studienbeginn waren 67,5% der Patienten in NYHA-Klasse II, 31,6% in Klasse III und 0,9% in Klasse IV eingestuft. Die mediane LVEF lag bei 32%. In beiden Behandlungsgruppen lag bei je 42% ein anamnestisch bekannter Typ-2-Diabetes mellitus vor, weitere 3% in jeder Gruppe wurden aufgrund eines sowohl zum Zeitpunkt der Aufnahme in die Studie als auch bei der Randomisierung gemessenen HbA1c ≥6,5% als Typ-2-Diabetiker klassifiziert.
Patienten mit einer schweren Einschränkung der Nierenfunktion (eGFR <30 ml/min/1.73 m2) durften nicht an der Studie teilnehmen. Die mittlere eGFR zum Studienbeginn war 66 ml/min/1,73 m2.
Kardiovaskuläre Mortalität und Verschlechterung der Herzinsuffizienz
Dapagliflozin 10 mg war Placebo in der Prävention von CV-bedingten Todesfällen und der Verschlechterung der Herzinsuffizienz überlegen und mit einem einheitlichen Behandlungseffekt auf den primären und die sekundären Endpunkte verbunden.
Dapagliflozin reduzierte die Häufigkeit des primären zusammengesetzten Endpunkts aus CV-bedingtem Tod, Hospitalisierung wegen Herzinsuffizienz und dringendem Arztbesuch wegen Herzinsuffizienz (HR 0,74 [95%-KI 0,65, 0,85]; p<0,0001). Die Anzahl der notwendigen Behandlungen pro Jahr (NNT) betrug 26 (95%-KI 18, 46). Die Ereigniskurven für Dapagliflozin und Placebo trennten sich früh und liefen über den Studienzeitraum hinweg weiter auseinander (Abbildung 1).
Abbildung 1: Zeit bis zum ersten Auftreten des Kompositums aus CV-bedingtem Tod, Hospitalisierung wegen Herzinsuffizienz und dringendem Arztbesuch wegen Herzinsuffizienz
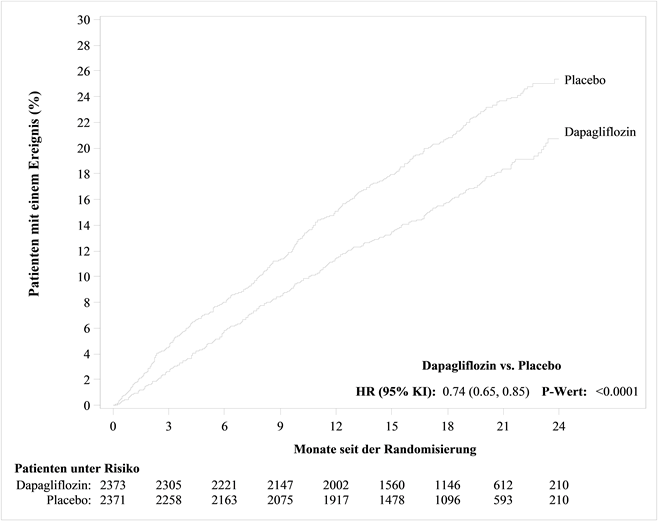
Ein dringender Arztbesuch wegen Herzinsuffizienz war definiert als dringende, nicht geplante Begutachtung durch einen Arzt, z.B. in einer Notaufnahme, mit bestehendem (über eine reine Erhöhung der Dosis oraler Diuretika hinausgehenden) Behandlungsbedarf wegen Verschlechterung der Herzinsuffizienz.
Patienten unter Risiko («patients at risk») sind diejenigen Patienten, die zu Beginn des Zeitraums noch am Leben waren.
Alle drei Komponenten des primären zusammengesetzten Endpunkts trugen zum Behandlungseffekt bei (Abbildung 2): Dapagliflozin reduzierte die Häufigkeit von kardiovaskulär bedingten Todesfällen und herzinsuffizienzbedingten Hospitalisationen (HR 0,75 [95%-KI 0,65; 0,85], p <0,0001).
Abbildung 2: Behandlungseffekte für den primären zusammengesetzten Endpunkt, dessen Komponenten und die Gesamtmortalität
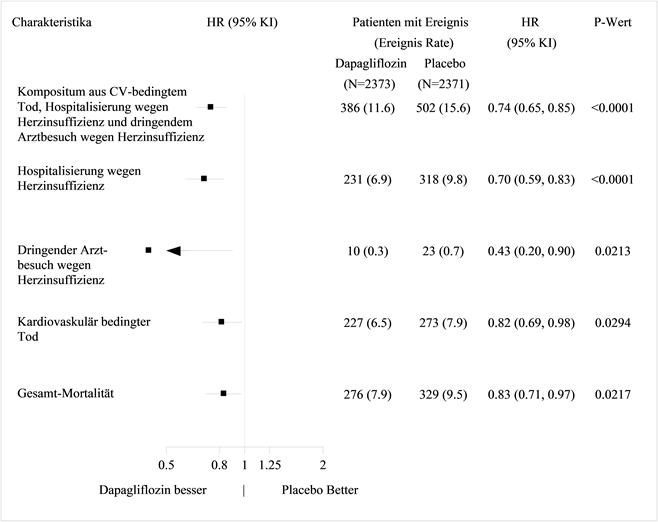
Ein dringender Arztbesuch wegen Herzinsuffizienz war definiert als dringende, nicht geplante Begutachtung durch einen Arzt, z.B. in einer Notaufnahme, mit bestehendem (über eine reine Erhöhung der Dosis oraler Diuretika hinausgehendem) Behandlungsbedarf wegen Herzinsuffizienzverschlechterung.
Die Anzahl der Erstereignisse für die einzelnen Komponenten repräsentiert die tatsächliche Anzahl der Erstereignisse für jede Komponente, und ihre Summe ist nicht gleich der Anzahl der Ereignisse im zusammengesetzten Endpunkt.
Die Ereignisraten sind als Anzahl der von dem Ereignis betroffenen Patienten pro 100 Patientenjahre der Beobachtung dargestellt.
Die p-Werte für die einzelnen Komponenten und die Gesamtmortalität sind nominal.
Der Nutzen der Behandlung mit Dapagliflozin war bei Herzinsuffizienz-Patienten sowohl mit als auch ohne Typ-2-Diabetes zu beobachten (Abbildung 3).
Abbildung 3: Behandlungseffekte bei allen Patienten, d.h. bei Patienten mit Typ-2-Diabetes und ohne Diabetes
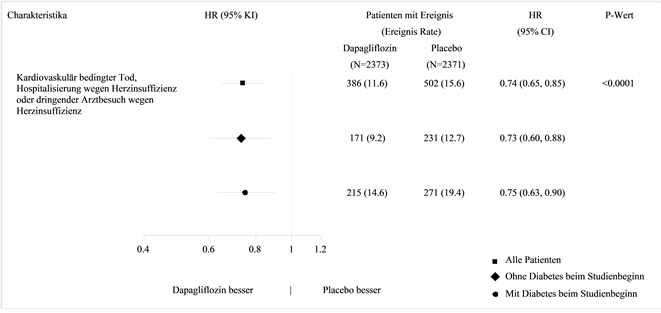
Ein dringender Arztbesuch wegen Herzinsuffizienz war definiert als dringende, nicht geplante Begutachtung durch einen Arzt, z.B. in einer Notaufnahme, mit bestehendem (über eine reine Erhöhung der Dosis oraler Diuretika hinausgehendem) Behandlungsbedarf wegen Verschlechterung der Herzinsuffizienz.
Die Ereignisraten sind als Anzahl der von dem Ereignis betroffenen Patienten pro 100 Patientenjahre der Beobachtung dargestellt
Der Nutzen der Behandlung mit Dapagliflozin gegenüber Placebo bezüglich des primären Endpunkts zeigte sich übereinstimmend auch in anderen wichtigen Subgruppen.
Pharmakokinetik
Absorption
Dapagliflozin wurde nach oraler Gabe schnell und gut absorbiert. Die maximalen Plasmakonzentrationen (Cmax) von Dapagliflozin wurden üblicherweise innerhalb von 2 Stunden nach Gabe im nüchternen Zustand erreicht. Nach einmal täglicher Gabe von Dosen von 10 mg betrugen die geometrischen Mittel der Cmax- und AUCτ-Werte von Dapagliflozin im steady state 158 ng/ml bzw. 628 ng h/ml. Die absolute orale Bioverfügbarkeit von Dapagliflozin nach Gabe einer 10-mg-Dosis beträgt 78%. Die Gabe zusammen mit einer fettreichen Mahlzeit verringerte die Cmax von Dapagliflozin um bis zu 50% und verlängerte tmax um ca. 1 Stunde, aber die AUC blieb verglichen mit dem Nüchternzustand unverändert. Diese Veränderungen werden nicht als klinisch bedeutsam angesehen.
Distribution
Dapagliflozin wird zu ungefähr 91% an Proteine gebunden. Die Proteinbindung war bei Vorliegen verschiedener Erkrankungen (z.B. Beeinträchtigung der Nieren- oder Leberfunktion) nicht verändert. Das mittlere Verteilungsvolumen von Dapagliflozin im steady state betrug 118 l.
Metabolismus
Dapagliflozin wird extensiv metabolisiert, wobei hauptsächlich der inaktive Metabolit Dapagliflozin-3-O-glucuronid entsteht. Dapagliflozin-3-O-glucuronid oder andere Metaboliten tragen nicht zur blutzuckersenkenden Wirkung bei. Die Bildung von Dapagliflozin-3-O-glucuronid wird über UGT1A9, ein Enzym, das in Leber und Niere vorkommt, vermittelt. Die CYP-vermittelte Metabolisierung war ein untergeordneter Abbauweg beim Menschen.
Elimination
Die mittlere terminale Halbwertszeit (t1/2) im Plasma betrug für Dapagliflozin 12,9 Stunden nach einer oralen Einzeldosis von 10 mg an gesunde Probanden. Dapagliflozin und dazugehörige Metaboliten werden hauptsächlich mit dem Urin ausgeschieden, davon weniger als 2% als unverändertes Dapagliflozin. Nach Verabreichung einer Dosis von 50 mg [14C]-Dapagliflozin wurden 96% wiedergefunden: 75% im Urin und 21% in den Fäzes. Über die Fäzes wurden etwa 15% der Dosis als unveränderter Wirkstoff ausgeschieden.
Linearität/Nicht Linearität
Die Dapagliflozin-Exposition erhöhte sich proportional zur Steigerung der Dapagliflozin-Dosis im Bereich von 0.1 bis 500 mg, und die Pharmakokinetik veränderte sich nicht im Laufe der Zeit bei wiederholter täglicher Dosierung über einen Zeitraum bis zu 24 Wochen.
Kinetik spezieller Patientengruppen
Eingeschränkte Leberfunktion
Bei Personen mit leichter oder moderater Leberfunktionsstörung (Child Pugh Klassen A und B) waren die mittleren Cmax und AUC-Werte von Dapagliflozin um bis zu 12% bzw. 36% höher als die der entsprechenden gesunden Kontrollpersonen. Diese Unterschiede wurden nicht als klinisch relevant erachtet. Bei Personen mit schwerer Leberfunktionsstörung (Child Pugh Klasse C) waren die mittleren Cmax und AUC-Werte von Dapagliflozin um 40% bzw. 67% höher als die der entsprechenden gesunden Kontrollpersonen.
Eingeschränkte Nierenfunktion
Im steady state (1x täglich 20 mg Dapagliflozin für 7 Tage) hatten Patienten mit Diabetes mellitus Typ 2 und leichter, moderater oder schwerer Nierenfunktionsstörung (bestimmt über die Iohexol-Plasmaclearance) mittlere systemische Dapagliflozin-Expositionen, die um 32%, 60% bzw. 87% höher waren als die von Patienten mit Diabetes mellitus Typ 2 und normaler Nierenfunktion. Der Einfluss einer Hämodialyse auf die Dapagliflozin-Exposition ist nicht bekannt.
Ältere Patienten
Bei Personen bis zu einem Alter von 70 Jahren gibt es keinen klinisch bedeutsamen Anstieg der Exposition, der allein auf dem Alter beruht. Jedoch kann eine erhöhte Exposition aufgrund der altersbedingten Abnahme der Nierenfunktion erwartet werden. Es gibt keine ausreichenden Daten, um Schlussfolgerungen in Bezug auf die Exposition bei Patienten >70 Jahre zu ziehen.
Kinder und Jugendliche
Die Pharmakokinetik bei Kindern und Jugendlichen wurde nicht untersucht.
Geschlecht
Die mittlere AUCss von Dapagliflozin bei Frauen (n=619) wurde in einem populationspharmakokinetischen Modell mit Daten aus Studien an gesunden Probanden und Patienten mit Diabetes um etwa 22% höher geschätzt als bei Männern (n=634; 90%-KI: 117%, 124%).
Körpergewicht
Die Dapagliflozin-Exposition nimmt mit steigendem Gewicht ab. Deshalb haben Patienten mit niedrigem Körpergewicht möglicherweise eine etwas erhöhte Exposition und Patienten mit hohem Körpergewicht eine etwas erniedrigte Exposition. Die Unterschiede in Bezug auf die Exposition wurden allerdings nicht als klinisch bedeutsam erachtet.
Ethnische Zugehörigkeit
Zwischen weissen, schwarzen oder asiatischen Bevölkerungsgruppen gab es keine klinisch relevanten Unterschiede in Bezug auf die systemische Exposition.
Präklinische Daten
Basierend auf konventionellen Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Genotoxizität, zum kanzerogenen Potenzial und zur Fertilität lassen die präklinischen Daten keine besonderen Gefahren für den Menschen erkennen.
Mutagenität
Dapagliflozin war in Ames-Mutagenitätstests negativ und in einer Reihe von in vitro Klastogenitätstests positiv in der Gegenwart von S9 Aktivierung und bei Konzentrationen ≥100 µg/ml. Dapagliflozin war bezüglich Klastogenität negativ in einer Reihe von in vivo Studien, in welchen die Reparatur des Mikronukleus oder der DNA bei Ratten bei Expositionen in Höhe des >2100-Fachen der klinischen Dosis evaluiert wurde.
Karzinogenität
In zweijährigen Kanzerogenitätsstudien induzierte Dapagliflozin über alle untersuchten Dosierungen hinweg weder an Ratten noch an Mäusen Tumore. In einer 6-Monatsstudie in Ratten, die mit 100 mg/kg N-Butyl-N-(4-hydroxybutyl)-Nitrosamin über 6 Wochen vorbehandelt wurden, wurde bei der Dosis von 0.5 mg/kg Dapagliflozin ein Anstieg der urothelialen Hyperplasien beobachtet. Im Vergleich zu den Tieren in der Kontrollgruppe wurde bei den mit Dapagliflozin behandelten Tieren keine Überrepräsentation maligner Läsionen verzeichnet.
Reproduktionstoxizität
Die direkte Gabe von Dapagliflozin an nicht mehr gesäugte Jungratten und die indirekte Exposition am Ende der Trächtigkeit (Zeitspannen entsprechend dem zweiten und dritten Schwangerschaftstrimester in Bezug auf die menschliche renale Reifung) und Stillzeit sind jeweils mit einer erhöhten Inzidenz und/oder Schwere von Dilatationen des Nierenbeckens und der Nierentubuli bei den Nachkommen verbunden.
Diese Expositionsphasen fallen in das kritische Fenster der renalen Reifung bei Ratten. Da die funktionelle Reifung der Nieren beim Menschen auch noch während der ersten 2 Lebensjahre stattfindet, könnten die bei juvenilen Ratten verzeichneten, Dapagliflozin-assoziierten renalen pelvischen und tubulären Dilatationen ein mögliches Risiko für die Reifung der Nieren beim Menschen während der ersten 2 Lebensjahre darstellen. Darüber hinaus sprechen die bei abgesetzten juvenilen Ratten nach Exposition während der Laktationsphase beobachteten ungünstigen Effekte auf die Zunahme des Körpergewichts dafür, dass Forxiga in den ersten 2 Lebensjahren nicht angewendet werden darf.
In einer Toxizitätsstudie mit Jungtieren, bei der junge Ratten postnatal von Tag 21 bis Tag 90 direkt eine Dapagliflozin-Dosis erhielten, wurde über Dilatationen des Nierenbeckens und der Nierentubuli über alle Dosisbereiche hinweg berichtet; die Welpen-Expositionen bei der niedrigsten getesteten Dosis entsprachen dem ≥15-Fachen der maximal empfohlenen Dosis für den Menschen. Diese Befunde waren verbunden mit dosisabhängigen Erhöhungen des Nierengewichts und einer makroskopischen Nierenvergrösserung, die über alle Dosen hinweg beobachtet wurden. Die bei Jungtieren beobachteten Dilatationen des Nierenbeckens und der Nierentubuli waren innerhalb der etwa 1-monatigen Genesungsperiode nicht vollständig reversibel.
In einer separaten Studie zur prä- und postnatalen Entwicklung erhielten trächtige Ratten ab Tag 6 der Tragzeit bis Tag 21 nach der Geburt Dosierungen, und die Welpen wurden der Substanz indirekt in utero und während der Stillzeit ausgesetzt. (Es wurde eine Satellitenstudie durchgeführt, um die Dapagliflozin-Expositionen in der Milch und in den Welpen zu bewerten.) Eine erhöhte Inzidenz oder Schwere von Dilatationen des Nierenbeckens wurde bei den erwachsenen Nachkommen der behandelten Muttertiere beobachtet, allerdings nur bei der höchsten getesteten Dosis (die damit verbundenen Dapagliflozin-Expositionen bei Muttertier und Welpen betrugen das 1415-Fache bzw. 137-Fache der Humanwerte bei der maximal empfohlenen Dosis für den Menschen). Eine zusätzliche Entwicklungstoxizität beschränkte sich auf eine dosisabhängige Gewichtsabnahme der Welpen und wurde nur bei einer Dosis von ≥15 mg/kg/Tag beobachtet (in Verbindung mit Welpen-Expositionen entsprechend dem 29-Fachen der Humanwerte bei der maximal empfohlenen Dosis für den Menschen). Eine maternale Toxizität war nur bei der getesteten Höchstdosis evident; sie war auf vorübergehende Reduktionen des Körpergewichts und der Nahrungsaufnahme beschränkt. Der no observed adverse effect level (NOAEL) für Entwicklungstoxizität, die niedrigste getestete Dosis, ist mit einem Vielfachen der maternalen systemischen Exposition verbunden, die etwa dem 19-Fachen der Humanwerte bei der für den Menschen empfohlenen Maximaldosis entspricht.
In zusätzlichen embryo-fetalen Entwicklungsstudien an Ratten und Kaninchen wurde Dapagliflozin in Intervallen gegeben, die mit den Hauptperioden der Organogenese bei den jeweiligen Spezies zusammenfielen. Über alle getesteten Dosierungen hinweg wurden am Kaninchen weder maternale noch Entwicklungstoxizitäten beobachtet; die getestete Höchstdosis ist mit einem Vielfachen der systemischen Exposition verbunden, die etwa dem 1'191-Fachen der für den Menschen empfohlenen Maximaldosis entspricht. Bei Ratten war Dapagliflozin weder embryoletal noch teratogen bei Expositionen bis zum 1'441-Fachen der für den Menschen empfohlenen Maximaldosis.
Fertilität
Bei männlichen und weiblichen Ratten zeigte Dapagliflozin bei keiner der untersuchten Dosen Auswirkungen auf die Fertilität.
Sonstige Hinweise
Beeinflussung diagnostischer Methoden
Interferenzen des Arzneimittels mit 1,5- anhydroglucitol -Assay
Die Überwachung der Glykämie mit 1,5-AG-Assays ist nicht empfohlen, weil bei Patienten, die mit SGLT2 Inhibitoren behandelt werden 1,5-AG-Messungen unzuverlässig zur Beurteilung der glykämischen Kontrolle sind. Alternative Methoden sollen zur Aufzeichnung der glykämischen Kontrolle benutzt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Nicht über 30 °C lagern.
Ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer
65176 (Swissmedic).
Zulassungsinhaberin
AstraZeneca AG, 6340 Baar.
Stand der Information
Juli 2020.
Composizione
Principi attivi
Dapagliflozin sotto forma di dapagliflozin propanediolo monoidrato.
Sostanze ausiliarie
Nucleo della compressa
Cellulosa microcristallina (E460i)
Lattosio (25 mg di lattosio nelle compresse rivestite con film da 5 mg. 50 mg di lattosio nelle compresse rivestite con film da 10 mg)
Crospovidone (E1202)
Diossido di silicio (E551)
Magnesio stearato (E470b)
Rivestimento della compressa
Alcool polivinilico (E1203)
Biossido di titanio (E171)
Macrogol 3350
Talco (E553b)
Ossido di ferro giallo (E172)
Forma farmaceutica e quantità di principio attivo per unità
Compresse rivestite con film da 5 mg o 10 mg di dapagliflozin.
Indicazioni/Possibilità d'impiego
Forxiga è indicato, assieme alla dieta e all'attività fisica, negli adulti (a partire dai 18 anni) con diabete mellito di tipo 2 non sufficientemente controllato:
- come monoterapia
- come terapia combinata add-on con altri medicamenti ipoglicemizzanti
- come terapia combinata iniziale con metformina
Per i risultati degli studi clinici rispetto alle associazioni con altri medicinali e agli effetti su eventi cardiovascolari vedere paragrafo «Efficacia Clinica».
Trattamento dell'insufficienza cardiaca con ridotta frazione d'eiezione (FEVS ≤40%, classe NYHA II-IV) ad integrazione di altre terapie farmacologiche dell'insufficienza cardiaca in pazienti adulti (vedere paragrafo «Efficacia clinica»).
Posologia/Impiego
Diabete mellito
Monoterapia e terapia combinata add-on
La dose iniziale raccomandata di Forxiga è di 5 mg 1 volta al giorno. Nei pazienti che tollerano Forxiga 5 mg al giorno e che necessitano di un maggiore controllo glicemico, la dose può essere aumentata a 10 mg al giorno.
Le compresse possono essere assunte indipendentemente dai pasti, a qualsiasi ora, e vanno deglutite intere.
Quando dapagliflozin viene utilizzato in combinazione con insulina o con un principio attivo insulinotropico, quale ad esempio una sulfonilurea, si può prendere in considerazione una dose inferiore di insulina o del principio attivo insulinotropico, per ridurre il rischio di ipoglicemia (vedere «Avvertenze e misure precauzionali»).
Terapia combinata iniziale
La posologia iniziale raccomandata di dapagliflozin nell'ambito di una terapia combinata iniziale con metformina corrisponde a 5 mg di dapagliflozin una volta al giorno.
Insufficienza cardiaca
La dose raccomandata di Forxiga per il trattamento dell'insufficienza cardiaca è di 10 mg una volta al giorno. L'assunzione può avvenire indipendentemente dai pasti in qualunque momento della giornata.
Forxiga può essere utilizzato in associazione ad altri medicamenti contro l'insufficienza cardiaca.
Pazienti con disturbi della funzionalità epatica
Nei pazienti con disturbo della funzionalità epatica di grado lieve o moderato, non è necessario alcun aggiustamento della dose. Nei pazienti con disturbo della funzionalità epatica di grado severo, si deve iniziare con una dose di 5 mg. Solo quando viene ben tollerata, la dose può essere aumentata a 10 mg (vedere «Avvertenze e misure precauzionali» e «Farmacocinetica»).
Pazienti con disturbi della funzionalità renale
Trattamento del diabete mellito
Nei pazienti con velocità di filtrazione glomerulare stimata (eGFR) persistentemente <45 ml/min/1,73 m2, Forxiga non dovrebbe essere usato, perché l'efficacia di dapagliflozin dipende dalla funzionalità renale (vedere «Avvertenze e misure precauzionali», «Effetti indesiderati», «Efficacia clinica» e «Farmacocinetica»).
Nello studio specifico sull'efficacia in pazienti con eGFR compresa tra 45 e <60 ml/min/1,73 m2 (vedere «Proprietà/effetti») è stata esaminata solo la dose da 10 mg al giorno. In pazienti con eGFR compresa tra 45 ml/min/1,73 m2 e <60 ml/min/1,73 m2, una terapia con dapagliflozin dovrebbe essere avviata alla dose di 10 mg/giorno. In pazienti che durante un trattamento con dapagliflozin mostrano una riduzione dell'eGFR a <60 ml/min/1,73 m2, la terapia può invece proseguire alla posologia corrente, sotto stretto monitoraggio metabolico.
In pazienti con disturbo della funzionalità renale di grado lieve, non è necessario alcun aggiustamento della dose.
La dose da 5 mg non è stata controllata in uno studio che ha esaminato in modo specifico l'efficacia in pazienti con eGFR di 45-60 ml/min/1,73 m2.
Trattamento dell'insufficienza cardiaca
Non sono necessari aggiustamenti della dose in base alla funzionalità renale. L'utilizzo di Forxiga non è raccomandato in pazienti con grave compromissione della funzionalità renale (eGFR <30 ml/min/1,73 m2) (vedere «Avvertenze e misure precauzionali»).
Pazienti anziani
In generale, non si raccomanda un aggiustamento della dose sulla base dell'età.
Bambini e adolescenti
La sicurezza e l'efficacia nei bambini e negli adolescenti non sono documentate. Non sono disponibili dati.
Controindicazioni
Ipersensibilità al principio attivo o a una qualsiasi delle sostanze ausiliarie di questo medicamento.
Avvertenze e misure precauzionali
In generale
Forxiga non dovrebbe essere usato nei pazienti con diabete mellito di tipo 1 o per il trattamento di una chetoacidosi diabetica.
Uso nei pazienti con compromissione della funzionalità epatica
Le esperienze di studi clinici sui pazienti con compromissione della funzionalità epatica sono limitate. Nei pazienti con disturbo della funzionalità epatica di grado severo, l'esposizione a dapagliflozin aumenta (vedere «Posologia/impiego» e «Farmacocinetica»). La sicurezza e l'efficacia di dapagliflozin nei pazienti con disturbi della funzionalità epatica di grado severo non sono state esaminate.
Uso nei pazienti con compromissione della funzionalità renale
Le esperienze con dapagliflozin in caso di grave compromissione della funzionalità renale (eGFR <30 ml/min/1,73 m2) o di insufficienza renale terminale (ESRD) sono limitate.
Trattamento del diabete mellito
L'efficacia di dapagliflozin dipende dalla funzionalità renale. Pertanto Forxiga non dovrebbe essere utilizzato in pazienti con eGFR costantemente <45 ml/min/1,73 m2 per il trattamento del diabete al fine di migliorare il controllo glicemico.
Monitoraggio della funzionalità renale
Il monitoraggio della funzionalità renale viene raccomandato come segue:
- prima dell'inizio della terapia con dapagliflozin
- almeno una volta all'anno nei pazienti con funzionalità renale normale
- almeno 2-4 volte all'anno nei pazienti con riduzione lieve o moderata della funzionalità renale (vedere anche «Posologia/impiego», «Effetti indesiderati», «Proprietà/effetti», «Farmacodinamica» e «Farmacocinetica»)
- prima dell'uso concomitante di dapagliflozin e di un medicamento che può compromettere la funzionalità renale; successivamente, controllo periodico.
Se l'eGFR cade durevolmente sotto i 45 ml/min/1,73 m2, si deve porre termine alla terapia con dapagliflozin.
Uso nei pazienti con rischio d'ipotensione
A causa del meccanismo d'azione, dapagliflozin induce la diuresi osmotica, il che può portare alla riduzione moderata della pressione arteriosa osservata negli studi clinici (vedere «Proprietà/effetti»), che può essere più marcata nei pazienti con glicemia molto alta.
In caso di malattie intercorrenti che possono condurre a una deplezione di volume (ad es. malattie gastrointestinali), si raccomanda un attento monitoraggio del volume (ad es. esame obiettivo, misurazione della pressione arteriosa, esami di laboratorio comprendenti gli elettroliti). Nei pazienti che sviluppano una deplezione di volume, si raccomanda una sospensione temporanea del trattamento con dapagliflozin fino a correggere la deplezione di volume (vedere «Effetti indesiderati»).
Chetoacidosi in pazienti con diabete mellito
Durante il trattamento con Forxiga e altri inibitori di SGLT2 sono stati osservati casi di chetoacidosi diabetica (CAD) gravi e talvolta pericolosi per la vita in pazienti con diabete mellito di tipo 1 e 2. Forxiga non è indicato per il trattamento di pazienti con diabete mellito di tipo 1.
Nei pazienti che in trattamento con Forxiga presentano sintomi quali nausea, vomito, inappetenza, dolori addominali, sete eccessiva, disturbi respiratori, esaurimento e confusione, si dovrebbe ricercare la presenza di una chetoacidosi mediante un test dei corpi chetonici, anche se il glucosio ematico è inferiore a 14 mmol/l (250 mg/dl). Nel sospetto di una chetoacidosi, Forxiga dovrebbe essere interrotto fino a un accertamento conclusivo.
I fattori che predispongono alla chetoacidosi comprendono una bassa riserva funzionale di cellule beta (ad es. dopo precedente pancreatite o intervento chirurgico al pancreas), ridotto apporto calorico, dieta povera di carboidrati, riduzione della dose di insulina o aumentato fabbisogno di insulina a causa di malattie intercorrenti (ad es. infezioni), interventi chirurgici o abuso di alcool. In questi casi, Forxiga dovrebbe essere utilizzato con cautela.
Sulla base di limitati dati provenienti da studi clinici, l'insorgenza di una chetoacidosi nei diabetici di tipo 1 sembra più frequente se questi vengono trattati con inibitori di SGLT-2. Pertanto, dapagliflozin non va usato nei pazienti con diabete mellito di tipo 1.
Infezioni genitali
In pazienti con infezioni genitali micotiche ricorrenti, si dovrebbe riconsiderare il trattamento con dapagliflozin per accertarsi che il beneficio della terapia sia superiore al rischio (vedere «Effetti indesiderati»).
Uso concomitante con medicamenti che possono causare ipoglicemie
L'insulina e i principi attivi insulinotropici, quali ad esempio le sulfoniluree, possono indurre ipoglicemia. Pertanto, con la somministrazione concomitante di Forxiga può essere necessaria una riduzione della dose di insulina o dei principi attivi insulinotropici al fine di ridurre il rischio di ipoglicemia (vedere «Effetti indesiderati»).
Gangrena di Fournier
Nel corso della sorveglianza del medicamento dopo l'introduzione sul mercato, in pazienti con diabete mellito trattati con inibitori di SGLT2, tra cui Forxiga, sono stati riportati casi di fascite necrotizzante del perineo (gangrena di Fournier), un'infezione necrotizzante molto rara, ma grave e potenzialmente pericolosa per la vita, che richiede un intervento chirurgico urgente. Sono stati interessati sia uomini che donne. Gli esiti gravi hanno compreso ricoveri ospedalieri, diversi interventi chirurgici e casi di decesso.
I pazienti in trattamento con Forxiga che presentano dolori o dolorabilità alla pressione, eritema o gonfiori nell'area genitale o perineale, come anche febbre e malessere, dovrebbero essere esaminati per la presenza di fascite necrotizzante. Se sussiste un sospetto corrispondente, va subito avviato un trattamento con antibiotici ad ampio spettro ed eventualmente un debridement chirurgico. Forxiga dovrebbe essere interrotto e sostituito con un'adeguata alternativa terapeutica, monitorando attentamente la glicemia.
Sostanze ausiliarie
Le compresse rivestite con film contengono lattosio. I pazienti affetti da intolleranza ereditaria al galattosio, deficit di lattasi o malassorbimento di glucosio-galattosio non dovrebbero assumere questo medicamento.
Interazioni
La metabolizzazione di dapagliflozin avviene primariamente attraverso la glucurono-coniugazione dipendente da UGT1A9.
Gli effetti del fumo, dell'alimentazione, dei preparati fitoterapeutici e del consumo di alcool sulla farmacocinetica di dapagliflozin non sono stati specificamente esaminati.
Interazioni farmacocinetiche
Studi in vitro
In studi in vitro, dapagliflozin non ha né inibito il citocromo P450 (CYP) 1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, né ha indotto il CYP1A2, il CYP2B6 o il CYP3A4. Pertanto non ci si deve attendere che dapagliflozin alteri la clearance metabolica di medicamenti somministrati in concomitanza, che vengono metabolizzati da questi enzimi.
Effetti di altri medicamenti su dapagliflozin
In studi di interazione su soggetti sani, in cui è stato principalmente utilizzato un disegno con somministrazione unica, la farmacocinetica di dapagliflozin non è stata alterata da: metformina, pioglitazone, sitagliptin, glimepiride, voglibose, idroclorotiazide, bumetanide, valsartan o simvastatina.
Rifampicina
In seguito a somministrazione concomitante con rifampicina (un induttore di diversi trasportatori attivi e di enzimi responsabili della metabolizzazione dei medicamenti), è stata osservata una riduzione del 22% dell'esposizione sistemica a dapagliflozin, che però non ha avuto alcun effetto clinicamente significativo sull'escrezione urinaria del glucosio nelle 24 ore. Pertanto, non si raccomanda alcun aggiustamento della dose.
Altri induttori
Non ci si attende alcun effetto clinicamente rilevante con altri induttori (ad es. carbamazepina, fenitoina, fenobarbital).
Acido mefenamico
In seguito all'uso concomitante di dapagliflozin e acido mefenamico (un inibitore UGT1A9), è stato osservato un aumento del 55% dell'esposizione sistemica a dapagliflozin, ma senza effetti clinicamente significativi sull'escrezione urinaria del glucosio nelle 24 ore. Non si raccomanda alcun aggiustamento della dose.
Effetti di dapagliflozin su altri medicamenti
In studi di interazione su soggetti sani, in cui è stato principalmente utilizzato un disegno con somministrazione unica, dapagliflozin non ha alterato la farmacocinetica di metformina, pioglitazone, sitagliptin, glimepiride, idroclorotiazide, bumetanide, valsartan, digossina (un substrato della P-gp) o warfarin (S-warfarin, un substrato del CYP2C9) o gli effetti anticoagulanti del warfarin misurati in base all'INR. La combinazione di una singola dose di dapagliflozin da 20 mg con simvastatina (un substrato di CYP3A4) ha causato un aumento dell'AUC della simvastatina del 19% e un aumento dell'AUC dell'acido simvastatinico del 31%. L'aumento dell'esposizione a simvastatina e all'acido simvastatinico viene classificato come non clinicamente rilevante.
Interazioni farmacodinamiche
Diuretici
Dapagliflozin può potenziare l'effetto diuretico dei diuretici e aumentare il rischio di disidratazione e di ipotensione (vedere «Avvertenze e misure precauzionali»).
Gravidanza/Allattamento
Gravidanza
Non sono disponibili dati sull'utilizzo di dapagliflozin durante la gravidanza. Studi sui ratti hanno mostrato una tossicità riguardante lo sviluppo dei reni nel periodo corrispondente al secondo e terzo trimestre di gravidanza nell'essere umano (vedere «Dati preclinici»). Pertanto, dapagliflozin non dovrebbe essere utilizzato nel secondo e nel terzo trimestre di gravidanza.
Se viene accertata una gravidanza, Forxiga dovrebbe essere interrotto.
Allattamento
Non è noto se nell'essere umano dapagliflozin e/o i suoi metaboliti passino nel latte materno. I dati disponibili sulla farmacodinamica/tossicologia negli studi sugli animali hanno dimostrato che dapagliflozin/i suoi metaboliti vengono escreti nel latte; inoltre, nella progenie allattata sono emersi effetti farmacologicamente mediati (vedere «Dati preclinici»). Non è possibile escludere un rischio per il neonato/lattante. Pertanto, Forxiga non deve essere utilizzato durante l'allattamento.
Fertilità
L'effetto di dapagliflozin sulla fertilità nell'essere umano non è stato esaminato.
Effetti sulla capacità di condurre veicoli e sull'impiego di macchine
Forxiga non ha effetti o ha effetti trascurabili sulla capacità di condurre veicoli o sulla capacità di utilizzare macchine. I pazienti dovrebbero essere informati che, se dapagliflozin viene impiegato in combinazione con una sulfonilurea o con insulina, sussiste il rischio di ipoglicemia.
Effetti indesiderati
Riassunto del profilo di sicurezza
Negli studi clinici con dapagliflozin sono stati trattati con dapagliflozin più di 15'000 pazienti con diabete di tipo 2 e più di 2000 pazienti con insufficienza cardiaca.
La valutazione primaria di sicurezza e tollerabilità è stata condotta in un'analisi aggregata predefinita di 13 studi a breve termine (fino a 24 settimane) controllati con placebo, con 2'360 soggetti trattati con dapagliflozin 10 mg e 2'295 trattati con placebo.
Le reazioni avverse più frequentemente segnalate in tutti gli studi clinici sono state le infezioni genitali.
Dapagliflozin 5 mg è stato inoltre valutato in un'analisi aggregata di 12 studi di breve durata controllati con placebo. Sono stati analizzati i dati di 1145 pazienti trattati con dapagliflozin 5 mg, di 1193 pazienti trattati con dapagliflozin 10 mg e di 1393 pazienti che assumevano placebo. Questi dati originano da studi in monoterapia e da studi sulla terapia combinata con altri antidiabetici orali.
Nello studio specificamente incentrato sugli esiti cardiovascolari (CV), su pazienti con diabete di tip 2 (DECLARE), 8'574 pazienti hanno ricevuto Forxiga 10 mg e 8'569 hanno ricevuto placebo per un tempo medio di esposizione di 48 mesi. In totale, ci sono stati 30'623 pazienti-anno di esposizione a Forxiga.
Nello studio sull'outcome cardiovascolare con dapagliflozin in pazienti con insufficienza cardiaca con ridotta frazione d'eiezione (DAPA-HF), 2368 pazienti sono stati trattati con dapagliflozin 10 mg e 2368 pazienti con placebo per una durata mediana di esposizione di 18 mesi. La popolazione di pazienti comprendeva pazienti con eGFR ≥30 ml/min/1,73 m2 e pazienti con diabete di tipo 2 e non diabetici.
Il profilo di sicurezza di dapagliflozin è risultato complessivamente coerente per tutte le indicazioni esaminate. Casi di chetoacidosi diabetica sono stati osservati esclusivamente in pazienti con diabete mellito.
Elenco degli effetti indesiderati
I seguenti effetti indesiderati sono stati identificati negli studi clinici controllati con placebo e in segnalazioni successive all'omologazione sul mercato. Nessuno di questi è risultato dose-dipendente. Gli effetti indesiderati riportati sotto sono classificati per frequenza e classi sistemico-organiche (MedDRA). Le indicazioni della frequenza si basano sulle seguenti categorie: molto comune (≥1/10), comune (≥1/100, <1/10); non comune (≥1/1000, <1/100); raro (≥1/10'000, <1/1000), molto raro (<1/10'000).
Infezioni ed infestazioni
Comune: vulvovaginite, balanite e infezioni genitali correlatea, infezioni delle vie urinarieb.
Molto raro: gangrena di Fournier (fascite necrotizzante del perineo).
Patologie del sistema emolinfopoietico
Comune: aumento dell'ematocrito.
Disturbi del metabolismo e della nutrizione
Molto comune: ipoglicemia (quando usato con SU o insulina).
Comune*: deplezione di volumec, dislipidemia.
Non comune: sete.
Raro: chetoacidosi diabeticad (vedere «Avvertenze e misure precauzionali»).
Patologie gastrointestinali
Non comune: stitichezza.
Patologie della cute e del tessuto sottocutaneo
Non comune: iperidrosi.
Non nota: eruzione cutaneae.
Patologie del tessuto muscoloscheletrico e del tessuto connettivo
Comune: mal di schiena.
Patologie renali e urinarie
Comune: disuria, poliuriaf.
Non comune: nicturia, urea ematica aumentata.
Patologie dell'apparato riproduttivo e della mammella
Non comune: prurito vulvovaginale.
a Vulvovaginite, balanite e infezioni genitali correlate includono diversi termini standard tra cui: infezione vulvovaginale e candidosi, balanopostite, balanite, candida, infezione e ascessi del pene, vaginite batterica, ascesso vulvare.
b Le infezioni delle vie urinarie includono diversi termini standard tra cui: infezioni del tratto urinario, cistite, pielonefrite, trigonite, uretrite, prostatite.
c La deplezione di volume include i seguenti termini standard: disidratazione, ipovolemia, ipotensione.
d Identificata nell'ampio studio sugli esiti CV condotto in pazienti con diabete di tipo 2. La frequenza si basa su tasso annuale.
e L'eruzione cutanea comprende i seguenti termini standard: eruzione cutanea, eruzione cutanea generalizzata, eruzione cutanea con prurito, eruzione cutanea maculare, eruzione cutanea maculopapulare, eruzione cutanea pustolosa, eruzione cutanea vescicolare, eruzione cutanea eritematosa.
f La poliuria comprende i seguenti termini standard: pollachiuria, poliuria, aumento dell'escrezione urinaria.
Descrizione di alcuni effetti collaterali
Chetoacidosi diabetica (CAD)
Nello studio DECLARE con dapagliflozin in pazienti con diabete di tipo 2, negli 8'754 pazienti che hanno ricevuto dapagliflozin 10 mg e negli 8'569 pazienti che hanno ricevuto placebo con un tempo medio di esposizione di 48 mesi, sono stati riportati eventi di CAD in 27 pazienti del gruppo trattato con dapagliflozin 10 mg e in 12 pazienti del gruppo placebo. Gli eventi si sono verificati uniformemente durante il periodo dello studio. Dei 27 pazienti con eventi CAD nel gruppo trattato con dapagliflozin, 22 erano in trattamento concomitante con insulina al momento dell'evento. I fattori precipitanti per la CAD sono stati quelli attesi in una popolazione con diabete mellito di tipo 2 (vedere paragrafo «Avvertenze e misure precauzionali»).
Nello studio DAPA-HF è stata segnalata chetoacidosi diabetica in 3 pazienti con diabete mellito di tipo 2 del gruppo trattato con dapagliflozin e in nessun paziente del gruppo placebo.
Ipoglicemia
La frequenza delle ipoglicemie è risultata in funzione del tipo di terapia di base utilizzata nel rispettivo studio.
Negli studi con dapagliflozin in monoterapia, come terapia add-on con metformina o come terapia add-on con sitagliptin (con o senza metformina), la frequenza degli eventi di ipoglicemia lieve nel trattamento fino a 102 settimane è risultata simile (<5%) tra i gruppi di trattamento, compreso il gruppo placebo. In tutti gli studi, si sono verificati occasionalmente eventi di ipoglicemia grave, con incidenze comparabili nei gruppi trattati con dapagliflozin e con placebo. Gli studi sulla terapia add-on con sulfonilurea e sulla terapia add-on con insulina hanno evidenziato tassi più elevati di ipoglicemia (vedere «Avvertenze e misure precauzionali»).
In uno studio sulla terapia add-on con glimepiride, alle settimane 24 e 48 eventi di ipoglicemia lieve sono stati riscontrati più frequentemente nel gruppo trattato con dapagliflozin 10 mg più glimepiride (rispettivamente 6,0% e 7,9%) e nel gruppo trattato con dapagliflozin 5 mg più glimepiride (rispettivamente 5,5% e 8,3%) rispetto al gruppo trattato con placebo più glimepiride (rispettivamente 2,1% e 2,1%).
In uno studio sulla terapia add-on con insulina sono stati riportati eventi di ipoglicemia grave sono stati riportati alla settimana 24 e alla settimana 104 rispettivamente nello 0,5% e nell'1,0% dei pazienti trattati con dapagliflozin 10 mg più insulina, alla settimana 24 nello 0,5% dei pazienti trattati con dapagliflozin 5 mg più insulina e alle settimane 24 e 104 nello 0,5% del gruppo trattato con placebo più insulina. Eventi di ipoglicemia lieve sono stati segnalati alle settimane 24 e 104 rispettivamente nel 40,3% e nel 53,1% dei pazienti trattati con dapagliflozin 10 mg più insulina, nel 43,4% e nel 52,8% dei pazienti trattati con dapagliflozin 5 mg più insulina e nel 34,0% e nel 41,6% dei pazienti che avevano ricevuto placebo più insulina.
Nello studio DECLARE con dapagliflozin non è stato osservato un aumentato rischio di eventi di ipoglicemia grave nella terapia con dapagliflozin rispetto a placebo. Eventi di ipoglicemia grave sono stati segnalati in 58 (0,7%) pazienti del gruppo trattato con dapagliflozin e in 83 (1,0%) pazienti del gruppo placebo.
Nello studio DAPA-HF il numero di eventi di ipoglicemia grave è risultato comparabile nei due bracci di trattamento con 4 pazienti (0,2%). Eventi di ipoglicemia grave si sono verificati solo in pazienti con diabete di tipo 2.
Vulvovaginite, balanite e infezioni genitali correlate
Casi di vulvovaginite, balanite e infezioni genitali correlate sono stati segnalati nel 5,5% e nello 0,6% dei pazienti trattati rispettivamente con dapagliflozin 10 mg e placebo. La maggior parte delle infezioni è stata di grado da lieve a moderato, causando in rari casi l'interruzione del trattamento con dapagliflozin, e i pazienti hanno risposto a un primo trattamento con una terapia standard. Queste infezioni sono state più frequenti nelle donne (8,4% e 1,2% rispettivamente per dapagliflozin e placebo). Nei pazienti con anamnesi corrispondente è risultata più probabile un'infezione ricorrente.
Nei dati aggregati dei 12 studi di breve durata controllati con placebo, vulvovaginite, balanite e altre infezioni genitali sono state riferite nello 0,9% dei pazienti trattati con placebo, nel 5,7% dei pazienti trattati con dapagliflozin 5 mg e nel 4,8% dei pazienti trattati con dapagliflozin 10 mg.
Nei pazienti con anamnesi corrispondente di infezioni genitali, è risultata più probabile un'infezione ricorrente.
Nell'ambito dello studio DAPA-HF, nel braccio trattato con dapagliflozin non sono stati riferiti casi di infezioni genitali gravi mentre nel gruppo placebo è stato registrato un caso di infezione genitale grave. A causa di un evento indesiderato dovuto a un'infezione genitale è stato necessario interrompere la terapia in sette pazienti (0,3%) del gruppo trattato con dapagliflozin e in zero pazienti del gruppo placebo.
Infezioni urinarie
Infezioni urinarie sono state riportate più frequentemente con dapagliflozin 10 mg rispetto a placebo (4,7% vs. 3,5%). La maggior parte delle infezioni è stata di grado da lieve a moderato, causando in rari casi l'interruzione del trattamento con dapagliflozin. Queste infezioni sono state più frequenti nelle donne, nei pazienti con anamnesi corrispondente è risultata più probabile un'infezione ricorrente. Occasionalmente è stata osservata una pielonefrite, che si è manifestata con frequenza simile a quella del trattamento di controllo.
Nei dati aggregati dei 12 studi di breve durata controllati con placebo, infezioni urinarie sono state riportate nel 3,7% dei pazienti trattati con placebo, nel 5,7% dei pazienti trattati con dapagliflozin 5 mg e nel 4,3% dei pazienti trattati con dapagliflozin 10 mg.
Nello studio DAPA-HF, il numero dei pazienti che presentavano una grave infezione urinaria è risultato basso e uniformemente distribuito: 14 pazienti (0,6%) del gruppo trattato con dapagliflozin e 17 pazienti (0,7%) del gruppo placebo. A causa di un evento indesiderato dovuto a un'infezione genitale è stato necessario interrompere la terapia in 5 pazienti (0,2%) del gruppo trattato con dapagliflozin e in 5 pazienti del gruppo placebo.
Effetti indesiderati renali
L'analisi dei 13 studi aggregati, controllati con placebo, di breve durata ha evidenziato un minimo aumento provvisorio della creatinina sierica nel gruppo trattato con dapagliflozin rispetto a placebo (differenza media dal basale alla settimana 1 e alla settimana 24: 0,.041 mg/dL versus 0,008 mg/dL e 0,019 mg/dL versus 0,008 mg/dL).
Esami di laboratorio
Ematocrito aumentato
Nei dati aggregati dei 13 studi controllati con placebo è stato osservato, nei pazienti che assumevano Forxiga, un aumento dell'ematocrito medio rispetto al basale, a partire dalla settimana 1 e fino alla settimana 16, in cui è stata rilevata la differenza media massima rispetto al basale. Alla settimana 24, la variazione media dell'ematocrito rispetto al basale è stata del 2,30% per dapagliflozin 10 mg versus -0,33% per placebo. Alla settimana 24 sono stati riportati valori di ematocrito >55% nell'1,3% dei pazienti trattati con dapagliflozin 10 mg e nello 0,4% dei pazienti trattati con placebo.
Valore aumentato del fosfato sierico inorganico
Alla settimana 24, nei dati aggregati dei 13 studi controllati con placebo, è stato osservato, nei pazienti trattati con dapagliflozin rispetto a placebo, un aumento del valore medio del fosfato sierico rispetto al basale (aumento medio 0,13 mg/dl versus -0,04 mg/dl). Alla settimana 24, l'1,7% dei pazienti trattati con Forxiga 10 mg ha presentato un'iperfosfatemia (≥5,6 mg/dl nel gruppo di età 17-65 anni e ≥5,1 mg/dl nel gruppo di età ≥66 anni) versus lo 0,9% dei pazienti trattati con placebo.
Effetti sui lipidi
Alla settimana 24, nei dati aggregati dei 13 studi controllati con placebo, la variazione percentuale media rispetto al basale per dapagliflozin 10 mg e per placebo è risultata essere la seguente: colesterolo totale 2,5% versus 0,0%; colesterolo HDL 6,0% versus 2,7%; colesterolo LDL 2,9% versus -1,0%; trigliceridi -2,7% versus -0,7%.
La notifica di effetti collaterali sospetti dopo l'omologazione del medicamento è molto importante. Consente una sorveglianza continua del rapporto rischio-beneficio del medicamento. Chi esercita una professione sanitaria è invitato a segnalare qualsiasi nuovo o grave effetto collaterale sospetto attraverso il portale online ElViS (Electronic Vigilance System). Maggiori informazioni sul sito www.swissmedic.ch.
Posologia eccessiva
Dopo singole dosi orali fino a 500 mg (50 volte superiori alla dose massima raccomandata nell'essere umano), dapagliflozin non ha mostrato alcuna tossicità. In questi soggetti è stata documentata, in maniera dose-dipendente, una glicosuria di differente durata (almeno per 5 giorni nel caso della dose da 500 mg) in assenza di disidratazione, ipotensione o variazioni degli elettroliti e di effetti clinicamente significativi sull'intervallo QTc. Nei pazienti, l'incidenza di ipoglicemia con dapagliflozin è risultata comparabile a placebo. Negli studi clinici in cui per 2 settimane sono state utilizzate, in soggetti sani e in pazienti con diabete mellito di tipo 2, dosi fino a 100 mg una volta al giorno (corrispondenti a 10 volte la dose massima raccomandata nell'essere umano), l'incidenza di ipoglicemia è risultata di poco più elevata rispetto a placebo e non dose-dipendente. La frequenza degli eventi indesiderati, comprese disidratazione o ipotensione, è risultata simile a quella con placebo e non vi sono state alterazioni dose-dipendenti, clinicamente significative, dei valori di laboratorio, inclusi elettroliti sierici e biomarker della funzionalità renale.
Trattamento
In caso di sovradosaggio, a seconda dello stato clinico del paziente dovrebbe essere avviato un trattamento di supporto appropriato. L'eliminazione di dapagliflozin mediante emodialisi non è stata esaminata.
Proprietà/Effetti
Codice ATC
A10BK01
Meccanismo d'azione
Dapagliflozin è un inibitore selettivo e reversibile del co-trasportatore sodio/glucosio 2 (SGLT2), che migliora il controllo glicemico nei pazienti con diabete mellito e assicura un beneficio cardio-renale.
L'inibizione di SGLT2 ottenuta con dapagliflozin riduce il riassorbimento del glucosio dal filtrato glomerulare nel tubulo renale prossimale limitando al contempo il riassorbimento del sodio, con conseguente escrezione del glucosio con l'urina e diuresi osmotica. Dapagliflozin aumenta pertanto l'afflusso di sodio al tubulo distale, il che presumibilmente determina un più marcato feedback tubulo-glomerulare e la riduzione della pressione intraglomerulare. Gli effetti secondari dell'inibizione di SGLT2 con dapagliflozin comprendono inoltre una lieve riduzione della pressione arteriosa, un calo ponderale e un aumento dell'ematocrito.
Dapagliflozin migliora i livelli plasmatici di glucosio a digiuno e nel postprandiale, riducendo il riassorbimento renale del glucosio e portando, quindi, all'escrezione urinaria del glucosio. Questa escrezione renale del glucosio si osserva già dopo la prima dose, persiste per l'intervallo di somministrazione di 24 ore e viene mantenuta per l'intera durata del trattamento.
La quantità di glucosio eliminata per via renale attraverso questo meccanismo dipende dalla concentrazione di glucosio ematico e dalla GFR. Quindi, in soggetti con livelli normali di glucosio, dapagliflozin presenta solo un basso potenziale di induzione di un'ipoglicemia. L'efficacia di dapagliflozin è indipendente dalla secrezione e dall'azione dell'insulina.
SGLT2 viene espresso selettivamente nel rene. Dapagliflozin non ha alcun effetto inibitorio su altri glucotrasportatori responsabili del trasporto del glucosio nei tessuti periferici. Dapagliflozin presenta una selettività per SGLT2 che è 1400 volte superiore rispetto a quella per SGLT1, il principale trasportatore responsabile del trasporto di glucosio nell'intestino.
Farmacodinamica
In soggetti sani e in pazienti con diabete mellito di tipo 2, dopo somministrazione di dapagliflozin è stato osservato un aumento della quantità di glucosio escreto nelle urine. In pazienti con diabete mellito di tipo 2, dopo 12 settimane di trattamento con dapagliflozin 10 mg al giorno, l'escrezione renale di glucosio è stata di 70 g al giorno. Il tasso massimo di eliminazione del glucosio è stato osservato a una dose di 20 mg/giorno di dapagliflozin. In pazienti con diabete mellito di tipo 2 che avevano ricevuto 10 mg di dapagliflozin al giorno per un periodo fino a 2 anni è emersa una glicosuria persistente.
Allo steady state, l'escrezione urinaria di glucosio nelle 24 ore è risultata dipendente in larga misura dalla funzionalità renale: Rispettivamente 85, 52, 18 o 11 g di glucosio al giorno sono stati escreti da pazienti con diabete mellito di tipo 2 e con funzionalità renale normale o disturbo della funzionalità renale di grado lieve, moderato o severo.
Nei pazienti con diabete mellito di tipo 2, la glicosuria indotta da dapagliflozin causa anche una diuresi osmotica e un volume urinario aumentato. L'aumento del volume urinario è risultato di circa 375 ml/giorno dopo un trattamento con 10 mg al giorno per 12 settimane. L'aumento del volume urinario è risultato associato a un aumento minimo e temporaneo dell'escrezione renale di sodio, che tuttavia non è stato accompagnato da variazioni della concentrazione sierica di sodio.
Anche l'escrezione renale di acido urico è risultata temporaneamente (per 3-7 giorni) aumentata e accompagnata da una persistente riduzione dei livelli sierici di acido urico. Dopo 24 settimane, la riduzione dei livelli sierici di acido urico era compresa tra -18,3 e -48,3 µmol/l (da -0,33 a -0,87 mg/dl).
Efficacia clinica
Diabete mellito
Per la valutazione dell'efficacia e della sicurezza di Forxiga, sono stati condotti 22 studi clinici in doppio cieco, randomizzati, controllati, su un totale di oltre 28'000 pazienti con diabete di tipo 2; in questi studi, oltre 15'000 pazienti sono stati trattati con dapagliflozin. Nella maggior parte degli studi, la durata del trattamento è stata di 24 settimane; nelle successive estensioni in aperto, i pazienti sono stati trattati per un periodo fino a 156 settimane.
L'effetto di dapagliflozin sugli eventi cardiovascolari in pazienti con diabete mellito di tipo 2 con o senza malattia cardiovascolare manifesta è stato esaminato in un ampio studio di esiti CV (DECLARE).
Controllo della glicemia
Monoterapia
Per valutare la sicurezza e l'efficacia di una monoterapia con dapagliflozin in pazienti con diabete mellito di tipo 2, è stato condotto uno studio in doppio cieco, controllato con placebo, della durata di 24 settimane (con un periodo di estensione supplementare). Il trattamento con dapagliflozin una volta al giorno ha determinato una riduzione statisticamente significativa della HbA1c rispetto a placebo (vedere tabella 1). Nel periodo di estensione, la riduzione della HbA1c è stata mantenuta fino alla settimana 102 (variazione media aggiustata rispetto al basale di -0,63%, -0,77% e -0,18% rispettivamente per dapagliflozin 10 mg, dapagliflozin 5 mg e placebo).
Tabella 1: Risultati alla settimana 24 (LOCFa) di uno studio controllato con placebo con dapagliflozin in monoterapia
Monoterapia | |||
|---|---|---|---|
Dapagliflozin | Dapagliflozin | Placebo | |
| Nb | 70 | 64 | 75 |
HbA1c (%) | |||
| Valore medio al basale | 8,01 | 7,83 | 7,79 |
| Variazione rispetto al basalec | -0,89 | -0,77 | -0,23 |
Differenza rispetto a placeboc (IC 95%) | -0,66* (-0,96; -0,36) | -0,54* (-0,84; -0,24) | |
Peso corporeo (kg) | |||
| Valore medio al basale | 94,13 | 87,17 | 88,77 |
| Variazione rispetto al basalec | -3,16 | -2,83 | -2,19 |
Differenza rispetto a placeboc (IC 95%) | -0,97 (-2,20; 0,25) | -0,65 (-1,90; 0,61) | |
a LOCF (last observation carried forward): ultimo valore disponibile per ciascun paziente (per i pazienti sottoposti a terapia di salvataggio, prima di tale terapia)
b Tutte le persone randomizzate che hanno assunto almeno una dose del trattamento in studio in doppio cieco durante la breve fase in doppio cieco
c Media dei minimi quadrati (LS), aggiustata in funzione del valore al basale
* Valore di p <0,0001 versus placebo
Terapia combinata
In uno studio di non inferiorità con controllo attivo, della durata di 52 settimane (con periodo di estensione di 52 settimane), dapagliflozin è stato valutato come terapia add-on alla metformina rispetto a una sulfonilurea (glipizide), come terapia add-on alla metformina, in persone con controllo glicemico insufficiente (HbA1c >6,5% e ≤10%). I risultati hanno mostrato una simile riduzione media della HbA1c dal basale alla settimana 52 rispetto a glipizide e confermano così la non-inferiorità (vedere tabella 2). Alla settimana 104, la variazione media aggiustata della HbA1c rispetto al basale è stata di -0,32% per dapagliflozin e di -0,14% per glipizide. Entro le settimane 52 e 104, almeno un evento ipoglicemico si è manifestato in una percentuale significativamente più bassa di pazienti del gruppo trattato con dapagliflozin (rispettivamente 3,5% e 4,3%) rispetto al gruppo trattato con glipizide (rispettivamente 40,8% e 47,0%). Alla settimana 104, nello studio erano rimasti ancora il 56,2% dei pazienti del gruppo trattato con dapagliflozin e il 50% dei pazienti del gruppo trattato con glipizide.
Tabella 2: Risultati alla settimana 52 (LOCFa) di uno studio con controllo attivo che ha confrontato dapagliflozin e glipizide come terapia add-on con metformina
Parametri | Dapagliflozin | Glipizide |
|---|---|---|
| Nb | 400 | 401 |
HbA1c (%) | ||
| Valore medio al basale | 7,69 | 7,74 |
| Variazione rispetto al basalec | -0,52 | -0,52 |
Differenza rispetto a glipizide + metforminac (IC 95%) | 0,00d (-0,11; 0,11) | |
Peso corporeo (kg) | ||
| Valore medio al basale | 88,44 | 87,60 |
| Variazione rispetto al basalec | -3,22 | 1,44 |
Differenza rispetto a glipizide + metforminac (IC 95%) | -4,65* (-5,14; -4,17) | |
a LOCF: ultimo valore disponibile per ciascun paziente
b Persone randomizzate e trattate per le quali era disponibile il valore al basale e con almeno 1 misurazione di efficacia dopo il basale
c Media dei minimi quadrati (LS), aggiustata in funzione del valore al basale
d Non inferiore rispetto a glipizide + metformina
* Valore di p <0,0001
Dapagliflozin come add-on a metformina, glimepiride, sitagliptin (con o senza metformina) o insulina ha determinato riduzioni statisticamente significative della HbA1c alla settimana 24 (p <0,0001; vedere tabelle da 3 a 6) rispetto ai pazienti che hanno ricevuto placebo.
In base ai dati a 48 settimane (glimepiride) e ai dati fino a 104 settimane (insulina), la riduzione della HbA1c osservata alla settimana 24 si è mantenuta negli studi sulla terapia combinata add-on (glimepiride e insulina). Nella terapia add-on con sitagliptin (con o senza metformina), la variazione media aggiustata rispetto al basale alla settimana 48 per dapagliflozin 10 mg e per placebo è stata rispettivamente pari a -0,30% e a 0,38%. La riduzione della HbA1c nell'ambito dello studio sulla terapia add-on con metformina si è mantenuta fino alla settimana 102 (variazione media aggiustata rispetto al basale, pari rispettivamente a -0,58%, -0,78% e 0,02% per 5 mg, 10 mg e placebo).
Nello studio con insulina (con o senza aggiunta di ipoglicemizzante orale), la variazione media aggiustata della HbA1c alla settimana 104 rispetto al basale è stata di ‑0,71% per dapagliflozin 5 mg, -0,71% per dapagliflozin 10 mg e -0,06% per placebo. Nelle persone trattate con dapagliflozin 5 mg o 10 mg, alle settimane 48 e 104, la dose di insulina a una dose media compresa nell'intervallo di 75-79 UI/giorno è rimasta stabile rispetto al basale. Nel gruppo placebo, l'aumento medio rispetto al basale è stato di 10,5 UI/giorno alla settimana 48 e di 18,3 UI/giorno alla settimana 104 (dose media di 84 UI/giorno e 92 UI/giorno). Alla settimana 104, nello studio erano rimasti ancora il 72,4% del gruppo trattato con dapagliflozin e il 54,8% del gruppo trattato con glipizide.
Tabella 3: Risultati degli studi controllati con placebo di 24 settimane (LOCFa) con dapagliflozin come terapia combinata add-on con metformina
Terapia combinata add-on | |||
|---|---|---|---|
Metformina¹ | |||
Dapagliflozin | Dapagliflozin | Placebo | |
| Nb | 135 | 137 | 137 |
HbA1c (%) | |||
| Valore medio al basale | 7,92 | 8,17 | 8,11 |
| Variazione rispetto al basalec | -0,84 | -0,70 | -0,30 |
Differenza rispetto a placeboc (IC 95%) | -0,54* (-0,74; -0,34) | -0,41* (-0,61; -0,21) | |
Persone (%), che raggiungono una HbA1c <7%: Aggiustato per il valore al basale | 40,6** | 37,5** | 25,9 |
Peso corporeo (kg) | |||
| Valore medio al basale | 86,28 | 84,73 | 87,74 |
| Variazione rispetto al basalec | -2,86 | -3,04 | -0,89 |
Differenza rispetto a placeboc (IC 95%) | -1,97* (-2,63; -1,31) | -2,16* (-2,81; -1,50) | |
1 Metformina ≥1500 mg/giorno
a LOCF: ultimo valore disponibile per ciascun paziente (per i pazienti sottoposti a terapia di salvataggio, prima di tale terapia)
b Tutte le persone randomizzate che hanno assunto almeno una dose del trattamento in studio in doppio cieco durante la breve fase in doppio cieco
c Media dei minimi quadrati (LS), aggiustata in funzione del valore al basale
* Valore di p <0,0001 versus placebo + ipoglicemizzante orale
** Valore di p <0,05 versus placebo + ipoglicemizzante orale
Tabella 4: Risultati degli studi controllati con placebo di 24 settimane (LOCFa) con dapagliflozin come terapia combinata add-on con glimepiride
Terapia combinata add-on | |||
Sulfonilurea (glimepiride1) | |||
Dapagliflozin 10 mg | Dapagliflozin 5 mg | Placebo | |
Nb | 151 | 142 | 145 |
HbA1c (%) | |||
Valore medio al basale | 8,07 | 8,12 | 8,15 |
Variazione rispetto al basalec | -0,82 | -0,63 | -0,13 |
Differenza rispetto a placeboc (IC 95%) | -0,68* (-0,86; -0,51) | -0,49* (-0,67; -0,32) | |
Persone (%), che raggiungono una HbA1c <7%: Aggiustato per il valore al basale | 31,7* | 30,3* | 13,0 |
Peso corporeo (kg) | |||
Valore medio al basale | 80,56 | 81,00 | 80,94 |
Variazione rispetto al basalec | -2,26 | -1,56 | -0,72 |
Differenza rispetto a placeboc (IC 95%) | -1,54* (-2,17; -0,92) | -0,84* (-1,47; -0,21) | |
1 Glimepiride 4 mg/giorno
a LOCF: ultimo valore disponibile per ciascun paziente (per i pazienti sottoposti a terapia di salvataggio, prima di tale terapia)
b Tutte le persone randomizzate che hanno assunto almeno una dose del trattamento in studio in doppio cieco durante la breve fase in doppio cieco
c Media dei minimi quadrati (LS), aggiustata in funzione del valore al basale
* Valore di p <0,0001 versus placebo + ipoglicemizzante orale
Tabella 5: Risultati degli studi controllati con placebo di 24 settimane (LOCFa) con dapagliflozin come terapia combinata add-on con sitagliptin (con o senza metformina)
Terapia combinata add-on | ||
Inibitore della DPP-4 (sitagliptin2) ± metformina1 | ||
Dapagliflozin 10 mg | Placebo | |
Nb | 223 | 224 |
HbA1c (%) | ||
Valore medio al basale | 7,90 | 7,97 |
Variazione rispetto al basalec | -0,45 | 0,04 |
Differenza rispetto a placeboc (IC 95%) | -0,48* (-0,62; -0,34) | |
Peso corporeo (kg) | ||
Valore medio al basale | 91,02 | 89,23 |
Variazione rispetto al basalec | -2,14 | -0,26 |
Differenza rispetto a placeboc (IC 95%) | -1,89* (-2,37; -1,40) | |
1 Metformina ≥1500 mg/giorno
2 Sitagliptin 100 mg/giorno
a LOCF: ultimo valore disponibile per ciascun paziente (per i pazienti sottoposti a terapia di salvataggio, prima di tale terapia)
b Tutte le persone randomizzate che hanno assunto almeno una dose del trattamento in studio in doppio cieco durante la breve fase in doppio cieco
c Media dei minimi quadrati (LS), aggiustata in funzione del valore al basale
* Valore di p <0,0001 versus placebo + ipoglicemizzante orale
Tabella 6: Risultati alla settimana 24 (LOCFa) di uno studio controllato con placebo con dapagliflozin in combinazione con insulina (da sola o con ipoglicemizzanti orali)
Parametri | Dapagliflozin 10 mg + insulina ± ipoglicemizzanti orali2 | Dapagliflozin 5 mg + insulina ± ipoglicemizzanti orali2 | Placebo + insulina ± ipoglicemizzanti orali2 |
Nb | 194 | 211 | 193 |
HbA1c (%) | |||
Valore medio al basale | 8,58 | 8,61 | 8,46 |
Variazione rispetto al basalec | -0,90 | -0,82 | -0,30 |
Differenza rispetto a placeboc (IC 95%) | -0,60* (-0,74; -0,45) | -0,52* (-0,66; -0,38) | |
Peso corporeo (kg) | |||
Valore medio al basale | 94,63 | 93,20 | 94,21 |
Variazione rispetto al basalec | -1,67 | -0,98 | 0,02 |
Differenza rispetto a placeboc (IC 95%) | -1,68* (-2,19; -1,18) | -1,00* (-1,50; -0,50) | |
Dose giornaliera media di insulina (UI)1 | |||
Valore medio al basale | 77,96 | 77,10 | 73,96 |
Variazione rispetto al basalec | -1,16 | -0,61 | 5,08 |
Differenza rispetto a placeboc (IC 95%) | -6,23* (-8,84; -3,63) | -5,69* (-8,25; -3,13) | |
a LOCF: ultimo valore disponibile per ciascun paziente (prima o il giorno della prima titolazione verso l'alto di insulina, se necessario)
b Tutte le persone randomizzate che hanno assunto almeno una dose del trattamento in studio in doppio cieco durante la breve fase in doppio cieco
c Media dei minimi quadrati (LS), aggiustata in funzione del valore al basale e della presenza di un ipoglicemizzante orale
* Valore di p <0,0001 versus placebo + insulina ± ipoglicemizzante orale
1 La titolazione verso l'alto del regime di insulina (inclusa insulina rapida, intermedia e basale) era consentita solo se le persone avevano soddisfatto criteri predefiniti di FPG.
2 Al basale, il 50% delle persone ha ricevuto insulina in monoterapia; il 50% ha ricevuto 1 o 2 ipoglicemizzanti orali in aggiunta a insulina: in quest'ultimo gruppo, l'80% ha ricevuto metformina da sola, il 12% ha ricevuto una terapia con metformina più sulfonilurea e il resto ha ricevuto altri ipoglicemizzanti orali.
Combinazione di dapagliflozin e dell'agonista del recettore GLP-1 (GLP-1-RA)
Uno studio randomizzato (1:1:1), in doppio cieco, a 3 bracci, della durata di 28 settimane ha esaminato l'effetto ipoglicemizzante della combinazione dapagliflozin (10 mg una volta al giorno) più un GLP-1-RA (exenatide retard 2 mg una volta la settimana) e delle monoterapie con il GLP-1-RA e con dapagliflozin. Lo studio ha incluso 694 pazienti adulti con diabete mellito di tipo 2 e insufficiente controllo glicemico (HbA1c ≥8,0 e ≤12,0%) in precedente monoterapia con metformina (≥1500 mg/giorno; proseguita senza variazioni durante la fase di trattamento). L'endpoint primario era la variazione della HbA1c da inizio studio alla settimana 28. La combinazione di dapagliflozin e GLP-1-RA è risultata superiore a entrambe le monoterapie nella riduzione della HbA1c (vedere tabella 7).
Tabella 7: Effetto ipoglicemizzante di dapagliflozin in combinazione con exenatide retard, dapagliflozin ed exenatide retard in pazienti trattati con metformina (fase di trattamento di 28 settimane)
Parametri | Dapagliflozin 10 mg QD + | Dapagliflozin 10 mg QD + | Exenatide retard 2 mg QW |
Nc | 228 | 230 | 227 |
HbA1c (%) | |||
Valore medio al basale | 9,29 | 9,25 | 9,26 |
Variazione rispetto al basalea | -1,98 | -1,39 | -1,60 |
Differenza rispetto al placebo (IC al 95%) | -0,59* (-0,84, -0,34) | ||
Differenza rispetto a exenatide retard QW (IC al 95%) | -0,38§ (-0,63, -0,13) | ||
Percentuale dei pazienti che hanno raggiunto una HbA1c <7,0%b | 44,7% | 19,1% | 26,9% |
QD = una volta al giorno, QW = una volta alla settimana, N = numero di pazienti nel gruppo di trattamento, IC = intervallo di confidenza.
a La modellazione delle medie aggiustate dei minimi quadrati (medie LS) e la(e) differenza(e) tra i gruppi di trattamento per la variazione dal basale alla settimana 28 si basano su un modello misto a misure ripetute (MMRM; mixed model with repeated measures) con trattamento, regione, stratificazione di HbA1c al basale (<9,0% o ≥9,0%), settimana e interazione trattamento × settimana come fattori fissi e con il valore al basale come covariata.
b Categorie ricavate da misurazioni continue. Per tutti i pazienti con dati sugli endpoint mancanti, imputazione come non-responder. Confronto dei trattamenti mediante test di Cochran-Mantel-Haenszel (CMH) stratificato secondo il valore di HbA1c al basale (<9,0% o ≥9,0%). Valori di p secondo la statistica associativa generale.
c Pazienti che hanno ricevuto almeno una volta il trattamento in studio e per i quali è disponibile almeno una misurazione di HbA1c post-basale.
* p <0,001.
§ p <0,01.
Tutti i valori di p sono aggiustati per la molteplicità.
Dalle analisi sono state escluse le misurazioni dopo terapia di salvataggio e dopo interruzione anticipata del trattamento in studio.
Terapia combinata iniziale con metformina
In due studi controllati a 3 bracci di 24 settimane l'efficacia e la sicurezza di un trattamento con metformina XR, dapagliflozin, metformina XR più dapagliflozin sono state esaminate in un totale di 1'236 pazienti naive alla terapia con diabete mellito di tipo 2 non sufficientemente controllato (HbA1c ≥7,5% e ≤12%). La dose di metformina XR è stata di 2000 mg in entrambi gli studi. La dose di dapagliflozin è stata di 5 mg e di 10 mg nei rispettivi studi. In tutti i tre bracci di trattamento di entrambi gli studi è stata osservata una riduzione significativa della HbA1c; la terapia combinata (metformina XR più dapagliflozin) in entrambi gli studi è risultata superiore alle monoterapie con metformina XR o con dapagliflozin (tabella 8).
Tabella 8: Risultati di due studi controllati con dapagliflozin nell'ambito di una terapia combinata iniziale con metformina XR (fase di trattamento di 24 settimane)
Parametri | Dapagliflozin + | Dapagliflozin + | Metformina XR + | |||
Posologia di dapagliflozin nello studio | 5 mg | 10 mg | 5 mg | 10 mg | 5 mg | 10 mg |
N† | 194 | 211 | 203 | 219 | 201 | 208 |
HbA1c (%) | ||||||
Valore medio al basale | 9,21 | 9,10 | 9,14 | 9,03 | 9,14 | 9,03 |
Variazione rispetto al basale (media aggiustata‡) | -2,05 | -1,98 | -1,19 | -1,45 | -1,35 | -1,44 |
Differenza rispetto a dapagliflozin (media aggiustata‡) (IC al 95%) | -0,86§ (-1,11,-0,62) | -0,53§ (-0,74, -0,32) | ||||
Differenza rispetto a metformina XR (media aggiustata‡) (IC al 95%) | -0,70§ (-0,94,-0,45) | -0,54§ (-0,75, -0,33) | -0,01¶ (-0,22, 0,20) | |||
Percentuale di pazienti che raggiungono una HbA1c <7,0%, aggiustata per il valore al basale | 52,4% | 46,6%# | 22,5% | 31,7% | 34,6% | 35,2% |
Variazione di HbA1c rispetto al basale in pazienti con HbA1c ≥9% al basale (media aggiustata‡) | -3,01 | -2,59# | -1,67 | -2,14 | -1,82 | -2,05 |
Differenza rispetto a dapagliflozin (media aggiustata‡) (IC al 95%) | -1,34§ (-1,72, -0,96) | -0,45# (-0,81, -0,09) | ||||
Differenza rispetto a metformina XR (media aggiustata‡) (IC al 95%) | -1,19§ (-1,57, -0,82) | -0,53 (-0,89, -0,18) | ||||
* LOCF: last observation carried forward: Per la valutazione finale è stato impiegato l'ultimo (prima dell'eventuale terapia di salvataggio) valore disponibile per ciascun paziente.
† Tutti i pazienti randomizzati che hanno assunto almeno una dose di trattamento in studio in doppio cieco durante la breve fase in doppio cieco.
‡ Media dei minimi quadrati aggiustata per il valore al basale.
§ Valore di p <0,0001.
¶ Non inferiore versus metformina XR.
# Valore di p <0,05.
Glucosio plasmatico a digiuno (fasting plasma glucose, FPG)
Il trattamento con dapagliflozin 5 mg o 10 mg in monoterapia o come terapia add-on con metformina, glimepiride o sitagliptin (con o senza metformina) ha determinato una riduzione statisticamente significativa del glucosio plasmatico a digiuno (da -1,90 a -1,18 mmol/l) rispetto a placebo (da ‑0,33 a 0,21 mmol/l). Questo effetto è stato osservato alla settimana 1 di trattamento e si è mantenuto negli studi condotti fono alla settimana 102.
Glucosio post-prandiale
Il trattamento con dapagliflozin 10 mg come terapia add-on con glimepiride ha determinato fino alla settimana 24 una riduzione statisticamente significativa del glucosio post-prandiale (2 ore dopo il pasto) che si è mantenuta fino alla settimana 48.
Il trattamento con dapagliflozin 10 mg come terapia add-on con sitagliptin (con o senza metformina) ha determinato fino alla settimana 24 una riduzione del glucosio post-prandiale (2 ore dopo il pasto), che si è mantenuta fino alla settimana 48.
Pressione arteriosa
Secondo l'analisi dei dati aggregati di 13 studi controllati con placebo, alla settimana 24 il trattamento con dapagliflozin 10 mg ha evidenziato una variazione della pressione arteriosa sistolica di -3,7 mmHg rispetto al basale e della pressione diastolica di -1.8 mmHg, versus ‑0,5 mmHg della sistolica e -0,5 mmHg della diastolica nel gruppo placebo. Questo effetto si è mantenuto fino alla settimana 104. Nei dati aggregati dei 12 studi di breve durata controllati con placebo, il trattamento con dapagliflozin 5 mg ha causato una variazione della pressione arteriosa sistolica di -3,5 mmHg rispetto al basale e della pressione diastolica di -2,1 mmHg.
Pazienti con disturbo della funzione renale
Disturbo della funzionalità renale di grado moderato (eGFR ≥45 fino a <60 ml/min/1,73 m2)
Uno studio in doppio cieco, randomizzato, controllato con placebo, ha esaminato l'efficacia di dapagliflozin su un totale di 321 pazienti adulti con diabete mellito di tipo 2 e una eGFR compresa tra ≥45 e <60 ml/min/1,73 m2 (cioè con disturbo della funzionalità renale di grado moderato corrispondente a CKD 3A) e insufficiente controllo glicemico con il trattamento corrente. I pazienti sono stati trattati con 10 mg di dapagliflozin (n=159) o con placebo (n=161). A inizio studio, l'HbA1c dei pazienti inclusi era mediamente pari allo 8,2%. Nell'endpoint primario della variazione dell'HbA1c rispetto al basale, alla settimana 24 dapagliflozin 10 mg ha evidenziato una superiorità statisticamente significativa rispetto a placebo. La variazione media della HbA1c rispetto al basale è stata di -0,37% per dapagliflozin e di -0,03% per placebo, corrispondente a una differenza tra le terapie di -0,34% (IC 95% -0,53, -0,15).
Il profilo di sicurezza di dapagliflozin è risultato in linea con quello di precedenti studi sulla popolazione generale di pazienti con diabete mellito di tipo 2. All'inizio del trattamento, l'eGFR media è diminuita nel gruppo dapagliflozin (dapagliflozin: -3,39 ml/min/1,73 m2, placebo: -0,90 ml/min/1,73 m2) ed è rimasta stabile durante le successive 24 settimane della fase di trattamento. A tre settimane dall'interruzione di dapagliflozin non si rilevavano più differenze rilevanti nella eGFR tra il gruppo con controllo attivo e il gruppo placebo.
In un precedente studio su complessivamente n=252 diabetici sono stati inoltre inclusi anche pazienti con eGFR compresa tra 30 e <45 ml/min/1,73 m2 (eGFR media in questo studio 45 ml/min/1,73 m2). Questo studio non ha potuto evidenziare un'efficacia per dapagliflozin né alla dose di 5 mg né alla dose di 10 mg (vedere «Posologia/impiego»).
Pazienti con HbA1c ≥9% al basale
In pazienti con HbA1c ≥9,0% al basale, il trattamento con dapagliflozin 10 mg ha determinato una riduzione statisticamente significativa della HbA1c alla settimana 24 sia in monoterapia (variazione media aggiustata rispetto al basale: -2,04% e 0,19% rispettivamente per dapagliflozin 10 mg e placebo) sia nel trattamento add-on con metformina (variazione media aggiustata rispetto al basale: -1,32% e -0,53% rispettivamente per dapagliflozin e placebo).
Studio sugli esiticardiovascolari (DECLARE)
L'effetto di un trattamento con dapagliflozin sugli eventi cardiovascolari è stato monitorato per un periodo di osservazione medio di 4,2 anni nell'ambito di uno studio internazionale, multicentrico, in doppio cieco che ha esaminato gli esiti cardiovascolari (DECLARE), nel quale i pazienti con diabete di tipo 2 (durata media della malattia 11,9 anni) sono stati randomizzati al trattamento con dapagliflozin (N=8582) o placebo (N=8578) in aggiunta alla terapia in atto. Nel corso dello studio è stato possibile aggiustare la terapia farmacologica del diabete e di altre comorbidità in base allo «standard of care (SoC)».
La media dell'HbA1c o dell'IMC a inizio studio è stata rispettivamente pari all'8,3% e a 32,1 kg/m2. La popolazione di studio, con età media pari a 63,9 anni, era costituita al 59,4% da pazienti senza malattia CV pregressa con i seguenti fattori di rischio: età ≥55 (uomini) o ≥60 anni (donne) e dislipidemia, ipertensione o fumo. Il restante 40,6% dei pazienti presentava già a inizio studio una malattia CV manifesta. Nel 10,0% dei pazienti è stata riscontrata un'insufficienza cardiaca in anamnesi. La eGFR media è risultata pari a 85,2 ml/min/1,73 m2 (solo il 7,4% dei pazienti presentava una eGFR <60ml/min/1,73 m2) mentre il 30,3% dei pazienti presentava albuminuria.
A inizio studio, l'82,0% dei pazienti ha ricevuto metformina per abbassare il glucosio ematico; altri trattamenti ipoglicemizzanti comprendevano insulina (40,9%), sulfoniluree (42,7%), inibitori della DPP4 (16,8%) e GLP-1-agonisti (4,4%). Per il trattamento delle comorbidità CV, i pazienti hanno ricevuto ACE-inibitori o antagonisti dei recettori dell'angiotensina 2 (81,3%), statine (75,0%), inibitori dell'aggregazione piastrinica (61,1%), acido acetilsalicilico (55,5%), beta-bloccanti (52,6%), calcio-antagonisti (34,9%), diuretici tiazidici (22,0%) e diuretici dell'ansa (10,5%).
I risultati dell'analisi primaria sono riportati nella tabella 9.
Endpoint | Dapagliflozin (N=8582) | Placebo (N=8578) | HR [IC 95] | ||
Pazienti con evento | Tasso di eventi (ogni 1000 pazienti-anno) | Pazienti con evento | Tasso di eventi (ogni 1000 pazienti-anno) | ||
MACE | 756 (8,8) | 22,6 | 803 (9,4) | 24,2 | 0,93 [0,84; 1,03] |
Morte CV | 245 (2,9) | 7,0 | 249 (2,9) | 7,1 | 0,98 [0,82; 1,17] |
Infarto miocardico | 393 (4,6) | 11,7 | 441 (5,1) | 13,2 | 0,89 [0,77; 1,01] |
Ictus | 235 (2,7) | 6,9 | 231 (2,7) | 6,8 | 1,01 [0,84; 1,21] |
Insufficienza cardiaca - Composito | 417 (4,9) | 12,2 | 496 (5,8) | 14,7 | 0,83 [0,73; 0,95] |
Ospedalizzazione per insufficienza cardiaca | 212 (2,5) | 6,2 | 286 (3,3) | 8,5 | 0,73 [0,61; 0,88] |
Morte CV | 245 (2,9) | 7,0 | 249 (2,9) | 7,1 | 0,98 [0,82; 1,17] |
Insufficienza cardiaca
Lo studio Dapagliflozin And Prevention of Adverse outcomes in Heart Failure (DAPA-HF) è stato uno studio internazionale, multicentrico, randomizzato, in doppio cieco, controllato con placebo condotto su un totale di 4744 pazienti con insufficienza cardiaca (classe funzionale NYHA [New York Heart Association] II-IV) con ridotta frazione d'eiezione (frazione d'eiezione ventricolare sinistra [FEVS] ≤40%) volto ad esaminare l'effetto di dapagliflozin in associazione alla terapia di base standard sulla mortalità CV e sul peggioramento dell'insufficienza cardiaca (ospedalizzazione per insufficienza cardiaca e visita medica urgente per insufficienza cardiaca).
I pazienti trattati con una terapia per l'insufficienza cardiaca corrispondente allo standard di cura (Tabella 10) sono stati assegnati a dapagliflozin 10 mg (N = 2373) o a placebo (N = 2371).
Tabella 10: Terapia concomitante per l'insufficienza cardiaca a inizio studio
Terapia concomitante per l'insufficienza cardiaca | Percentuale di pazienti a inizio studio (%) |
ACE-inibitore o antagonista del recettore dell'angiotensina o inibitore del recettore dell'angiotensina-neprilisina (ARNI) | 94% |
ARNI | 11% |
Beta-bloccanti | 96% |
Antagonisti del recettore dei mineralcorticoidi (MRA) | 71% |
Diuretico | 93% |
Dispositivo impiantabile | 26% |
L'obiettivo primario dello studio era verificare se dapagliflozin consentisse di evitare decessi per cause cardiovascolari e il peggioramento dell'insufficienza cardiaca e di migliorare i sintomi dell'insufficienza cardiaca.
Il tempo di osservazione mediano è stato di 18 mesi. La popolazione di pazienti aveva un'età media di 66 anni ed era al 77% di sesso maschile; i pazienti erano per il 70% caucasici, per il 5% neri o afro-americani e per il 24% asiatici.
A inizio studio, il 67,5% dei pazienti risultava di classe NYHA II, il 31,6% di classe NYHA III e lo 0,9% di classe NYHA IV. La FEVS mediana è risultata pari al 32%. In entrambi i gruppi di trattamento, il 42% dei pazienti presentava diabete mellito di tipo 2 in anamnesi, un ulteriore 3% in ciascun gruppo è stato classificato con diabete di tipo 2 in ragione di un livello di HbA1c ≥6,5% sia al momento dell'inclusione nello studio sia alla randomizzazione.
Allo studio non hanno potuto partecipare pazienti con grave compromissione della funzionalità renale (eGFR <30 ml/min/1,73 m2). L'eGFR media a inizio studio è risultata pari a 66 ml/min/1,73 m2.
Mortalità cardiovascolare e peggioramento dell'insufficienza cardiaca
Per quanto riguarda la prevenzione dei decessi per cause CV e del peggioramento dell'insufficienza cardiaca, dapagliflozin 10 mg è risultato superiore al placebo e associato a un effetto del trattamento uniforme sull'endpoint primario e sugli endpoint secondari.
Dapagliflozin ha ridotto la frequenza dell'endpoint primario composito di morte per cause CV, ospedalizzazione per insufficienza cardiaca e visita medica urgente per insufficienza cardiaca (HR 0,74 [IC 95% 0,65, 0,85]; p <0,0001). Il numero dei trattamenti necessari su base annuale (NNT) è risultato pari a 26 (IC 95% 18, 46). Le curve degli eventi per dapagliflozin e placebo hanno evidenziato rapidamente un discostamento, che è aumentato ulteriormente durante il periodo dello studio (Figura 1).
Figura 1: Tempo fino alla prima comparsa del composito di morte per cause CV, ospedalizzazione per insufficienza cardiaca e visita medica urgente per insufficienza cardiaca
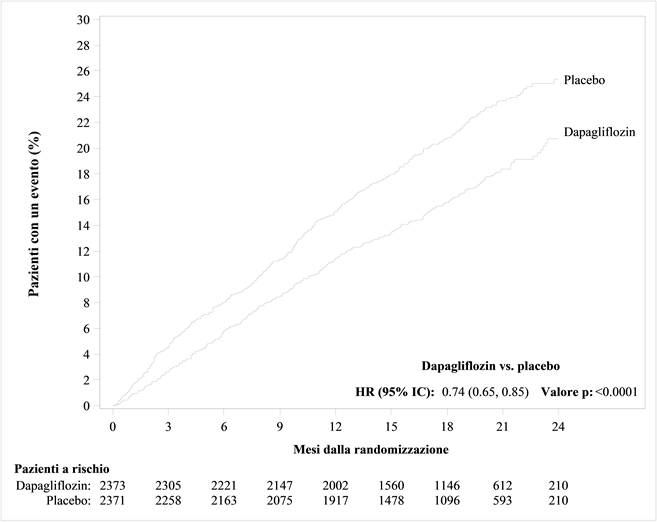
È stata definita visita medica urgente per insufficienza cardiaca un esame urgente, non programmato, da parte di un medico, ad es. in un pronto soccorso, con necessità corrente di trattamento (che esulava dal mero aumento della dose dei diuretici orali) a causa del peggioramento dell'insufficienza cardiaca.
I pazienti a rischio («patients at risk») sono i pazienti ancora in vita nel periodo considerato.
Tutti i tre componenti dell'endpoint primario composito hanno contribuito all'effetto del trattamento (Figura 2): dapagliflozin ha ridotto la frequenza dei decessi per cause cardiovascolari e delle ospedalizzazioni per insufficienza cardiaca (HR 0,75 [IC 95% 0,65; 0,85], p <0,0001).
Figura 2: Effetti del trattamento per l'endpoint primario composito, i suoi componenti e la mortalità totale
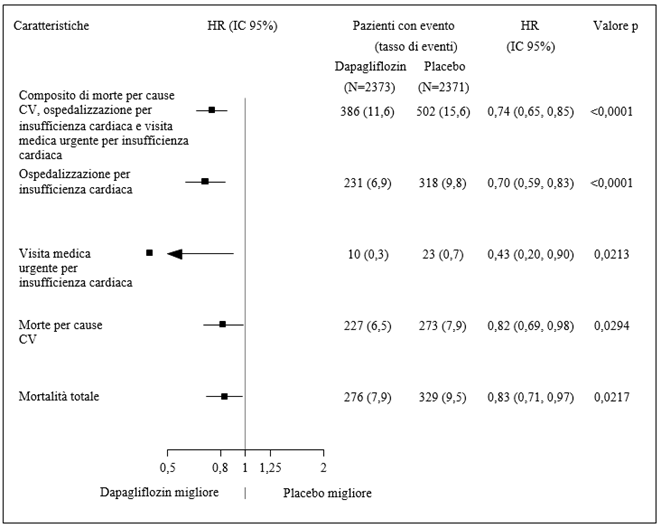
È stata definita visita medica urgente per insufficienza cardiaca un esame urgente, non programmato, da parte di un medico, ad es. in un pronto soccorso, con necessità corrente di trattamento (che esulava dal mero aumento della dose dei diuretici orali) a causa del peggioramento dell'insufficienza cardiaca.
Il numero dei primi eventi per i singoli componenti rappresenta il numero effettivo dei primi eventi per ogni componente, e la loro somma non coincide con il numero degli eventi nell'endpoint composito.
I tassi di eventi sono rappresentati come numero di pazienti interessati dall'evento ogni 100 anni-paziente di osservazione.
I valori p per i singoli componenti e la mortalità totale sono nominali.
Il beneficio del trattamento con dapagliflozin è stato osservato nei pazienti con insufficienza cardiaca sia con che senza diabete mellito di tipo 2 (Figura 3).
Figura 3: Effetti del trattamento in tutti i pazienti, ovvero con diabete di tipo 2 e senza diabete
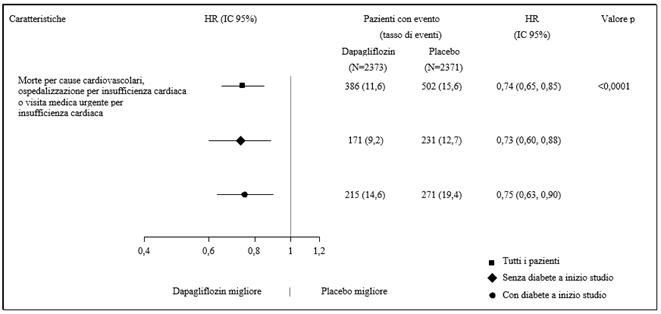
È stata definita visita medica urgente per insufficienza cardiaca un esame urgente, non programmato, da parte di un medico, ad es. in un pronto soccorso, con necessità corrente di trattamento (che esulava dal mero aumento della dose dei diuretici orali) a causa del peggioramento dell'insufficienza cardiaca.
I tassi di eventi sono rappresentati come numero di pazienti interessati dall'evento ogni 100 anni-paziente di osservazione.
Per quanto riguarda l'endpoint primario, il beneficio del trattamento con dapagliflozin rispetto al placebo è risultato coerente anche in altri importanti sottogruppi.
Farmacocinetica
Assorbimento
Dapagliflozin è stato assorbito rapidamente e bene dopo somministrazione orale. Le concentrazioni plasmatiche massime (Cmax) di dapagliflozin sono state raggiunte di regola entro 2 ore dalla somministrazione a digiuno. Dopo somministrazione una volta al giorno di dosi di 10 mg, le medie geometriche dei valori della Cmax e dell'AUC di dapagliflozin allo steady state sono risultate rispettivamente di 158 ng/ml e di 628 ng h/ml. La biodisponibilità orale assoluta di dapagliflozin dopo somministrazione di una dose di 10 mg è del 78%. La somministrazione assieme a un pasto ricco di grassi ha ridotto la Cmax di dapagliflozin fino al 50% e ha prolungato la tmax di circa 1 ora, ma la AUC è rimasta inalterata rispetto allo stato a digiuno. Queste variazioni non vengono considerate clinicamente significative.
Distribuzione
Dapagliflozin è legato alle proteine per circa il 91%. Il legame con le proteine non è risultato alterato in presenza di diverse malattie (ad es. compromissione della funzionalità renale o epatica). Il volume di distribuzione medio di dapagliflozin allo steady state è stato di 118 l.
Metabolismo
Dapagliflozin viene ampiamente metabolizzato, dando origine principalmente al metabolita inattivo dapagliflozin-3-O-glucuronide. Dapagliflozin-3-O-glucuronide o altri metaboliti non contribuiscono all'effetto ipoglicemizzante. La formazione di dapagliflozin-3-O-glucuronide viene mediata da UGT1A9, un enzima presente nel fegato e nei reni. La metabolizzazione mediata dal CYP è una via di degradazione secondaria nell'essere umano.
Eliminazione
L'emivita terminale media (t1/2) nel plasma di dapagliflozin è risultata pari a 12,9 ore dopo una singola dose orale di 10 mg in soggetti sani. Dapagliflozin e i suoi metaboliti vengono eliminati principalmente con l'urina, di cui meno del 2% come dapagliflozin immodificato. Dopo somministrazione di una dose di 50 mg di [14C]-dapagliflozin, è stato ritrovato il 96%: il 75% nell'urina e il 21% nelle feci. Con le feci viene eliminato circa il 15% della dose sotto forma di principio attivo immodificato.
Linearità/non linearità
L'esposizione a dapagliflozin è aumentata in maniera proporzionale all'aumentare della dose di dapagliflozin nell'intervallo da 0,1 a 500 mg, e la farmacocinetica non è cambiata nel tempo in seguito a somministrazione giornaliera ripetuta per un periodo fino a 24 settimane.
Cinetica di gruppi di pazienti speciali
Disturbi della funzionalità epatica
Nelle persone con disturbo della funzionalità epatica di grado lieve o moderato (classi Child Pugh A e B), i valori medi della Cmax e della AUC di dapagliflozin sono risultati più elevati, con aumento rispettivamente fino al 12% e al 36%, rispetto ai corrispondenti controlli sani. Queste differenze non sono state considerate clinicamente rilevanti. Nelle persone con disturbo della funzionalità epatica di grado severo (classi Child Pugh C), i valori medi della Cmax e della AUC di dapagliflozin sono risultati più elevati del 40% e del 67% rispetto ai corrispondenti controlli sani.
Disturbi della funzionalità renale
Allo steady state (20 mg di dapagliflozin una volta al giorno per 7 giorni), i pazienti con diabete mellito di tipo 2 e disturbo della funzionalità renale di grado lieve, moderato o severo (determinata mediante clearance plasmatica dello ioexolo) hanno presentato esposizioni sistemiche medie a dapagliflozin superiori rispettivamente del 32%, 60% e 87% a quella dei pazienti con diabete mellito di tipo 2 e funzionalità renale normale. L'effetto dell'emodialisi sull'esposizione a dapagliflozin non è noto.
Pazienti anziani
Nelle persone fino a un'età di 70 anni non è presente alcun aumento clinicamente significativo dell'esposizione basato unicamente sull'età. Tuttavia, è possibile attendersi un'aumentata esposizione a causa della compromissione della funzionalità renale correlata all'età. Non sono disponibili dati sufficienti per trarre conclusioni relative all'esposizione nei pazienti di età >70 anni.
Bambini e adolescenti
La farmacocinetica nei bambini e negli adolescenti non è stata esaminata.
Sesso
In un modello di farmacocinetica di popolazione con dati ricavati da studi su soggetti sani e su pazienti con diabete, la AUCss media di dapagliflozin nelle donne (n=619) è stata stimata superiore del 22% circa rispetto a quella degli uomini (n=634; IC al 90%: 117%, 124%).
Peso corporeo
L'esposizione a dapagliflozin diminuisce all'aumentare del peso corporeo. Pertanto è possibile che i pazienti con basso peso corporeo presentino un'esposizione un po' aumentata e i pazienti con peso corporeo elevato un'esposizione un po' ridotta. Tuttavia, le differenze relative all'esposizione non sono state considerate clinicamente significative.
Appartenenza etnica
Non sono emerse differenze clinicamente rilevanti nell'esposizione sistemica tra gruppi di popolazione caucasica, nera o asiatica.
Dati preclinici
I dati preclinici degli studi convenzionali su farmacologia di sicurezza, tossicità per somministrazione ripetuta, genotossicità, potenziale cancerogeno e fertilità non evidenziano alcun rischio particolare per l'essere umano.
Mutagenicità
Dapagliflozin è risultato negativo al test di mutagenicità di Ames e positivo a una serie di test di clastogenicità in vitro, in presenza di attivazione S9 e a concentrazioni ≥100 µg/ml. Dapagliflozin è risultato negativo per la clastogenicità in una serie di studi in vivo, in cui è stata valutata la riparazione del micronucleo o del DNA nei ratti a esposizioni >2100 volte superiori alla dose clinica.
Cancerogenicità
Negli studi di cancerogenicità a due anni, a tutte le posologie esaminate, dapagliflozin non ha indotto tumori né nei ratti né nei topi. In uno studio di 6 mesi su ratti precedentemente trattati con 100 mg/kg di N-butil-N-(4-idrossibutil)-nitrosammina per 6 settimane, è stato osservato un aumento delle iperplasie uroteliali con dapagliflozin alla dose di 0,5 mg/kg. Rispetto agli animali del gruppo di controllo, negli animali trattati con dapagliflozin non è stato riscontrato un aumento delle lesioni maligne.
Tossicità per la riproduzione
La somministrazione diretta di dapagliflozin a giovani ratti non più allattati e l'esposizione indiretta al termine della gravidanza (periodo corrispondente al secondo e terzo trimestre di gestazione per quanto riguarda la maturità renale umana) e durante l'allattamento sono correlate a un aumento dell'incidenza e/o della severità di dilatazioni della pelvi renale e dei tubuli renali nella progenie.
Queste fasi di esposizione cadono nella finestra critica della maturazione renale nei ratti. Poiché la maturazione funzionale dei reni nell'essere umano continua anche durante i primi 2 anni di vita, le dilatazioni della pelvi e dei tubuli renali associate a dapagliflozin e osservate nei giovani ratti potrebbero rappresentare un possibile rischio per la maturazione dei reni nell'essere umano nei primi 2 anni di vita. Inoltre, gli effetti avversi sull'aumento del peso corporeo, osservati in ratti giovani svezzati dopo esposizione nella fase di allattamento, depongono a favore del fatto che Forxiga non deve essere utilizzato nei primi 2 anni di vita.
In uno studio di tossicità con giovani animali, in cui giovani ratti avevano ricevuto direttamente una dose di dapagliflozin nel periodo post-natale dal giorno 21 al giorno 90, sono state riportate dilatazioni della pelvi e dei tubuli renali a tutti gli intervalli di dose; le esposizioni dei cuccioli alla dose più bassa esaminata corrispondevano a ≥15 volte la dose massima raccomandata nell'essere umano. Questi risultati sono stati associati ad aumenti dose-dipendenti del peso dei reni e a un ingrossamento macroscopico dei reni, osservati a tutte le dosi. Le dilatazioni della pelvi e dei tubuli renali osservate nei giovani animali non sono state completamente reversibili nel periodo di recupero di circa 1 mese.
In uno studio separato sullo sviluppo pre- e post-natale, ratte gravide sono state trattate a partire dal giorno 6 di gestazione e fino al giorno 21 post-partum, e i cuccioli sono stati indirettamente esposti alla sostanza in utero e durante l'allattamento. (È stato condotto uno studio satellite, per valutare le esposizioni a dapagliflozin nel latte e nei cuccioli). Un aumento dell'incidenza o della severità delle dilatazioni della pelvi renale è stata osservata nella progenie adulta di madri trattate, tuttavia soltanto alla dose massima testata (le relative esposizioni a dapagliflozin nelle madri e nei cuccioli erano rispettivamente 1415 volte e 137 volte superiori ai valori umani alla dose massima raccomandata nell'essere umano). Un'ulteriore tossicità per lo sviluppo si è limitata a una riduzione dose-dipendente del peso dei cuccioli ed è stata osservata solo a una dose ≥15 mg/kg/giorno (in associazione con l'esposizione dei cuccioli corrispondente a 29 volte i valori umani alla dose massima raccomandata nell'essere umano). Una tossicità materna è stata evidente solo alla dose massima testata e limitata a riduzioni transitorie del peso corporeo e dell'assunzione di cibo. Il no observed adverse effect level (NOAEL) per la tossicità per lo sviluppo, la dose più bassa testata, è correlato a un multiplo dell'esposizione sistemica materna, che corrisponde a circa 19 volte i valori umani alla dose massima raccomandata nell'essere umano.
In altri studi sullo sviluppo embrio-fetale nei ratti e nei conigli, dapagliflozin è stato somministrato a intervalli che coincidevano con i periodi principali dell'organogenesi nelle rispettive specie. A tutte le posologie testate, non è stata osservata tossicità materna o per lo sviluppo nel coniglio; la dose massima testata è correlata a un multiplo dell'esposizione sistemica, che corrisponde a circa 1'191 volte la dose massima raccomandata nell'essere umano. Nei ratti, dapagliflozin non è risultato né embrioletale né teratogeno a esposizioni fino a 1'441 volte la dose massima raccomandata nell'essere umano.
Fertilità
In ratti maschi e femmine, dapagliflozin non ha mostrato effetti sulla fertilità a nessuna delle dosi esaminate.
Altre indicazioni
Influenza su metodi diagnostici
Interferenze del medicamento con il dosaggio dell'1,5-anidroglucitolo
Il monitoraggio della glicemia mediante dosaggi dell'1,5-AG non è raccomandato, perché nei pazienti trattati con inibitori di SGLT2, le misurazioni dell'1,5-AG non sono affidabili per la valutazione del controllo glicemico. Per monitorare il controllo glicemico, vanno usati metodi alternativi.
Stabilità
Il medicamento non deve essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Indicazioni particolari concernenti l'immagazzinamento
Non conservare a temperature superiori a 30 °C.
Conservare fuori dalla portata dei bambini.
Numero dell'omologazione
65176 (Swissmedic).
Titolare dell’omologazione
AstraZeneca AG, 6340 Baar.
Stato dell'informazione
Luglio 2020.
Composition
Principes actifs
Dapagliflozine sous forme de dapagliflozine propanediol monohydraté.
Excipients
Noyau du comprimé
Cellulose microcristalline (E460i)
Lactose (25 mg de lactose dans les comprimés pelliculés à 5 mg. 50 mg de lactose dans les comprimés pelliculés à 10 mg)
Crospovidone (E1202)
Dioxyde de silicium (E551)
Stéarate de magnésium (E470b)
Pelliculage
Poly(alcool vinylique) (E1203)
Dioxyde de titane (E171)
Macrogol 3350
Talc (E553b)
Oxyde de fer jaune (E172)
Forme pharmaceutique et quantité de principe actif par unité
Comprimés pelliculés à 5 mg ou 10 mg de dapagliflozine.
Indications/Possibilités d’emploi
Forxiga est indiqué en complément d'un régime alimentaire et de l'exercice physique chez les adultes (à partir de 18 ans) atteints de diabète de type 2 insuffisamment contrôlé:
- En monothérapie
- En traitement adjuvant associé avec d'autres hypoglycémiants
- En traitement associé initial avec la metformine
Pour les résultats des études concernant les associations de traitements et les effets sur les évènements cardiovasculaires, voir la section « Efficacité clinique ».
Traitement de l'insuffisance cardiaque à fraction d'éjection réduite (FEVG ≤40%, classe NYHA II-IV) en complément d'autres traitements médicamenteux de l'insuffisance cardiaque chez les patients adultes (voir la section «Efficacité clinique»).
Posologie/Mode d’emploi
Diabète
Monothérapie et traitement adjuvant associé
La dose initiale recommandée de Forxiga est de 5 mg 1× par jour. La dose peut être augmentée à 10 mg par jour lorsque Forxiga 5 mg par jour est bien toléré et qu'un contrôle glycémique accru est nécessaire. Les comprimés peuvent être pris à tout moment de la journée, indépendamment des repas. Les comprimés doivent être avalés entiers.
Lorsque la dapagliflozine est utilisée en association avec l'insuline ou un sécrétagogue d'insuline, comme par exemple une sulfonylurée, une dose plus faible d'insuline ou de sécrétagogue d'insuline peut être envisagée pour réduire le risque d'hypoglycémie (voir «Mises en garde et précautions»).
Traitement associé initial
La dose initiale quotidienne recommandée de dapagliflozine en cas d'utilisation dans le cadre d'un traitement associé initial avec la metformine est de 5 mg.
Insuffisance cardiaque
La dose recommandée de Forxiga pour le traitement de l'insuffisance cardiaque est de 10 mg une fois par jour. Le médicament peut être pris indépendamment des repas, à tout moment de la journée.
Forxiga peut être utilisé conjointement avec d'autres médicaments contre l'insuffisance cardiaque.
Patients présentant des troubles de la fonction hépatique
Aucun ajustement de la posologie n'est nécessaire chez les patients atteints d'insuffisance hépatique légère ou modérée. Chez les patients atteints d'insuffisance hépatique sévère, la dose initiale recommandée est de 5 mg. La dose peut être augmentée à 10 mg lorsque le traitement est bien toléré (voir «Mises en garde et précautions» et «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
Traitement du diabète
Forxiga ne doit pas être utilisé chez les patients dont le taux de filtration glomérulaire estimé (TFGe) est constamment <45 ml/min/1,73 m2, étant donné que l'efficacité de la dapagliflozine dépend de la fonction rénale (voir «Mises en garde et précautions», «Effets indésirables», «Efficacité clinique» ainsi que «Pharmacocinétique»).
Dans l'étude d'efficacité spécifique menée chez des patients présentant un TFGe de 45 à <60 ml/min/1,73 m2 (voir «Propriétés / Effets»), seule la dose de 10 mg par jour a été examinée. Chez les patients présentant un TFGe entre 45 ml/min/1,73 m2 et <60 ml/min/1,73 m2, un traitement par la dapagliflozine doit être instauré à une dose de 10 mg/jour. Chez les patients chez lesquels une diminution du TFGe atteignant une valeur de <60 ml/min/1,73 m2 est observée au cours d'un traitement par la dapagliflozine, le traitement peut par contre être poursuivi à la posologie utilisée jusqu'à présent et sous contrôle étroit du métabolisme.
Aucun ajustement de la dose n'est nécessaire chez les patients atteints d'un trouble léger de la fonction rénale.
La dose de 5 mg n'a pas été examinée dans l'étude portant spécifiquement sur l'efficacité chez les patients présentant un TFGe de 45-60 ml/min/1,73 m2.
Traitement de l'insuffisance cardiaque
Il n'est pas nécessaire d'ajuster la posologie compte tenu de la fonction rénale. L'utilisation de Forxiga n'est pas recommandée chez les patients atteints d'insuffisance rénale sévère (TFGe <30 ml/min/1,73 m2) (voir «Mises en garde et précautions»).
Patients âgés
En général, aucun ajustement de la posologie n'est recommandé selon l'âge.
Enfants et adolescents
La tolérance et l'efficacité n'ont pas été établies chez les enfants et les adolescents. Aucune donnée n'est disponible.
Contre-indications
Hypersensibilité à la substance active ou à l'un des excipients du médicament.
Mises en garde et précautions
Générales
Forxiga ne doit pas être utilisé chez les patients atteints de diabète de type 1 ou pour le traitement de l'acidocétose diabétique.
Utilisation chez les patients présentant des troubles de la fonction hépatique
L'expérience des essais cliniques chez les patients atteints d'insuffisance hépatique est limitée. L'exposition à la dapagliflozine est augmentée chez les patients atteints d'insuffisance hépatique sévère (voir «Posologie/Mode d'emploi» et «Pharmacocinétique»). La sécurité et l'efficacité de la dapagliflozine n'ont pas été étudiées chez les patients atteints d'insuffisance hépatique sévère.
Utilisation chez les patients présentant des troubles de la fonction rénale
L'expérience portant sur la dapagliflozine chez les patients atteints d'insuffisance rénale sévère (TFGe <30 ml/min/1,73 m2) ou d'insuffisance rénale terminale (IRT) est limitée.
Traitement du diabète
L'efficacité de la dapagliflozine est dépendante de la fonction rénale. Par conséquent, Forxiga ne doit pas être utilisé pour le traitement du diabète en vue d'améliorer le contrôle glycémique chez les patients dont le TFGe est constamment <45 ml/min/1,73 m2.
Surveillance de la fonction rénale
La surveillance de la fonction rénale est recommandée comme suit:
- avant le début du traitement par la dapagliflozine.
- au moins une fois par an chez les patients présentant une fonction rénale normale.
- au moins 2 à 4 fois par an chez les patients présentant altération de la fonction rénale légère ou modérée (voir également «Posologie/Mode d'emploi», «Effets indésirables», «Propriétés/Effets», «Pharmacodynamique» et «Pharmacocinétique»).
- avant l'utilisation concomitante de la dapagliflozine et d'un médicament qui peut affecter la fonction rénale; ensuite, contrôle périodique.
Le traitement par la dapagliflozine doit être arrêté si le TFGe est constamment inférieur à 45 ml/min/1,73 m2.
Utilisation chez les patients à risque d'hypotension
En raison de son mécanisme d'action, la dapagliflozine induit la diurèse osmotique qui peut mener à une baisse modérée de la pression artérielle ainsi qu'observée pendant les études cliniques (voir «Propriétés/Effets»). Celle-ci pourrait être plus prononcée chez les patients ayant des glycémies très élevées.
Acidocétose chez les patients diabétiques
Des cas graves d'acidocétose diabétique (ACD), certains pouvant engager le pronostic vital, ont été rapportés chez des patients atteints de diabète de type 1 et de type 2 et traités par Forxiga et d'autres inhibiteurs du SGLT2. Forxiga ne doit pas être utilisé chez les patients atteints de diabète de type 1.
Chez les patients présentant sous Forxiga des symptômes tels que nausées, vomissements, anorexie, douleurs abdominales, soif intense, difficultés respiratoires, épuisement et confusion, il faut immédiatement rechercher une acidocétose au moyen d'un test de corps cétonique, même si la glycémie est inférieure à 14 mmol/l (250 mg/dl). Lorsqu'une acidocétose est suspectée, le traitement doit être arrêté jusqu'à la clarification finale de la situation.
Les facteurs prédisposant à l'acidocétose incluent une faible réserve de cellules bêta fonctionnelles (p.ex. antécédent de pancréatite ou d'intervention chirurgicale du pancréas), absorption réduite de calories, une alimentation pauvre en hydrates de carbone, réduction de la dose d'insuline ou besoin accru en insuline en raison d'affections intercurrentes (p.ex. infections), interventions chirurgicales ou abus d'alcool. Dans ces cas, Forxiga doit être utilisé avec prudence.
Sur la base de données limitées issues d'études cliniques, une acidocétose semble survenir plus fréquemment chez les diabétiques de type 1 lorsque ceux-ci sont traités par des inhibiteurs du SGLT2. Par conséquent, la dapagliflozine ne doit pas être utilisée chez les patients atteints de diabète de type 1.
Infections génitales
Chez les patients souffrant de mycoses génitales récidivantes, le traitement par dapagliflozine doit être reconsidéré pour s'assurer que le bénéfice du traitement est supérieur au risque encouru (voir «Effets indésirables»).
Utilisation concomitante de médicaments pouvant entraîner des hypoglycémies
L'insuline et les sécrétagogues d'insuline, comme les sulfonylurées, peuvent entraîner une hypoglycémie. Une dose plus faible d'insuline ou des sécrétagogues d'insuline peut donc être nécessaire lors de traitement concomitant avec Forxiga afin de réduire le risque d'hypoglycémie (voir «Effets indésirables»).
Gangrène de Fournier
Dans le cadre de la pharmacovigilance post-commercialisation, des cas de fasciite nécrosante du périnée (gangrène de Fournier) ont été rapportés chez des patients atteints de diabète prenant des inhibiteurs du SGLT2, y compris Forxiga. Cette infection nécrosante très rare, mais grave et mettant potentiellement en jeu le pronostic vital nécessite une intervention chirurgicale en urgence. Cette infection touchait aussi bien les femmes que les hommes. Les issues graves comprenaient des hospitalisations, plusieurs interventions chirurgicales et des décès.
Les patients qui sont traités par Forxiga et présentent des symptômes tels que des douleurs, une sensibilité à la pression, un érythème ou des tuméfactions dans la zone génitale ou périnéale, accompagnés de fièvre et de malaises doivent être examinés à la recherche d'une fasciite nécrosante. En cas de suspicion de gangrène de Fournier, un traitement immédiat par des antibiotiques à large spectre et éventuellement un débridement chirurgical doivent être instaurés. Le traitement par Forxiga doit être arrêté et remplacé par un autre traitement approprié. Dans ce contexte, la glycémie doit être étroitement surveillée.
Excipients
Les comprimés pelliculés contiennent du lactose. Les patients atteints de troubles héréditaires rares d'intolérance au galactose, de déficit en Lapp lactase ou de malabsorption du glucose-galactose ne devraient pas prendre ce médicament.
Interactions
Le métabolisme de la dapagliflozine se fait essentiellement via une réaction de glucuronoconjugaison médiée par l'UGT1A9.
Les effets du tabagisme, du régime alimentaire, des produits de phytothérapie et de la consommation d'alcool sur la pharmacocinétique de la dapagliflozine n'ont pas été étudiés.
Interactions pharmacocinétiques
Études in vitro
Lors d'études in vitro, la dapagliflozine n'a ni inhibé les cytochromes P450 (CYP) 1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, ni induit les CYP1A2, CYP2B6 ou CYP3A4. La dapagliflozine ne devrait ainsi pas modifier la clairance métabolique des médicaments coadministrés et métabolisés par ces enzymes.
Effet des autres médicaments sur la dapagliflozine
Les études d'interaction menées chez des sujets sains, principalement à dose unique, suggèrent que la pharmacocinétique de la dapagliflozine n'est pas modifiée par la metformine, la pioglitazone, la sitagliptine, le glimépiride, le voglibose, l'hydrochlorothiazide, le bumétanide, le valsartan ou la simvastatine.
Rifampincine
Suite à la coadministration de la dapagliflozine avec la rifampicine (un inducteur de différents transporteurs actifs et d'enzymes responsables du métabolisme de médicaments), une baisse de 22% de l'exposition systémique à la dapagliflozine a été observée, mais qui n'avait pas d'effet cliniquement significatif sur l'excrétion urinaire de glucose sur 24 heures. Aucun ajustement posologique n'est donc recommandé.
Autres inducteurs
Aucun effet cliniquement pertinent avec d'autres inducteurs (p.ex. carbamazépine, phénytoïne, phénobarbital) n'est attendu.
Acide méfénamique
Suite à la coadministration de la dapagliflozine avec l'acide méfénamique (un inhibiteur de l'UGT1A9), une augmentation de 55% de l'exposition systémique de la dapagliflozine a été observée, mais sans effet cliniquement significatif sur l'excrétion urinaire de glucose sur 24 heures. Aucun ajustement posologique n'est donc recommandé.
Effet de la dapagliflozine sur d'autres médicaments
Lors d'études d'interactions menées chez des sujets sains, principalement à dose unique, la dapagliflozine n'a pas modifié la pharmacocinétique de la metformine, de la pioglitazone, de la sitagliptine, du glimépiride, de l'hydrochlorothiazide, du bumétanide, du valsartan, de la digoxine (un substrat de la P-gp) ou de la warfarine (S-warfarine, un substrat du CYP2C9) ou des effets anticoagulants de la warfarine mesurés par l'INR. L'association d'une seule dose de dapagliflozine 20 mg et de simvastatine (un substrat du CYP3A4) a entraîné une augmentation de 19% de l'ASC de la simvastatine et de 31% de l'ASC de la simvastatine acide. L'augmentation de l'exposition à la simvastatine et à la simvastatine acide n'est pas considérée cliniquement significative.
Interactions pharmacodynamiques
Diurétiques
La dapagliflozine peut renforcer l'effet diurétique des diurétiques et peut augmenter le risque de déshydratation et d'hypotension (voir «Mises en garde et précautions»).
Grossesse/Allaitement
Grossesse
Il n'existe pas de données concernant l'emploi de la dapagliflozine chez la femme enceinte. Des études chez le rat ont révélé une toxicité pour le rein en développement durant la période correspondant aux deuxième et troisième trimestres de la grossesse humaine (voir «Données précliniques»). Par conséquent, l'utilisation de la dapagliflozine n'est pas recommandée au cours des deuxième et troisième trimestres de grossesse.
Le traitement par la dapagliflozine doit être interrompu dès qu'une grossesse est constatée.
Allaitement
On ne sait pas si la dapagliflozine et/ou ses métabolites sont excrétés dans le lait humain. Les données pharmacodynamiques/toxicologiques disponibles chez l'animal ont mis en évidence une excrétion de la dapagliflozine/de ses métabolites dans le lait, ainsi que des effets pharmacologiques induits par l'allaitement de la descendance (voir «Données précliniques»). Un risque pour le nouveau-né/nourrisson ne peut être exclu. Par conséquent, Forxiga ne doit pas être utilisé durant l'allaitement.
Fertilité
L'effet de la dapagliflozine sur la fertilité n'a pas été étudié chez l'être humain.
Effet sur l’aptitude à la conduite et l’utilisation de machines
Forxiga n'a pas d'effet ou qu'un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. Les patients doivent être informés du risque d'hypoglycémie lorsque la dapagliflozine est administrée en association avec des sulfonylurées ou de l'insuline.
Effets indésirables
Résumé du profil de sécurité
Dans les études cliniques sur la dapagliflozine, plus de 15 000 patients atteints de diabète de type 2 et plus de 2000 patients atteints d'insuffisance cardiaque ont été traités par dapagliflozine. L'évaluation primaire de la sécurité et de la tolérance a été effectuée lors d'une analyse poolée déterminée au préalable de 13 études à court terme (jusqu'à 24 semaines) contrôlées contre placebo, au cours desquelles 2360 personnes ont été traitées par dapagliflozine 10 mg et 2295 par placebo.
Les effets indésirables les plus fréquents rapportés dans toutes les études étaient des infections de la région génitale.
La dapagliflozine 5 mg a également été évaluée dans une analyse poolée de 12 études à court terme contrôlées contre placebo. L'analyse a porté sur les données de 1145 patients sous dapagliflozine 5 mg, 1193 patients sous dapagliflozine 10 mg resp. 1393 patients sous placebo. Ces données sont issues d'études en monothérapie et en combinaison avec d'autres antidiabétiques oraux.
Dans l'étude qui était spécifiquement consacrée aux issues cardiovasculaires (CV) chez les patients avec diabète de type 2 (DECLARE), 8574 patients ont reçu Forxiga 10 mg et 8569 patients un placebo pendant une durée d'exposition médiane de 48 mois. L'exposition à Forxiga a porté sur 30'623 patients-années au total.
Dans l'étude sur la dapagliflozine qui était consacrée aux bénéfices cardiovasculaires chez les patients atteints d'insuffisance cardiaque à fraction d'éjection réduite (DAPA-HF), 2368 patients ont été traités par dapagliflozine 10 mg et 2368 patients par placebo pendant une durée d'exposition médiane de 18 mois. La population de patients comprenait des patients présentant un TFGe ≥30 ml/min/1,73 m2 ainsi que des diabétiques de type 2 et des non-diabétiques.
Le profil de sécurité de la dapagliflozine était dans l'ensemble cohérent dans tous les domaines d'application étudiés. Des cas d'acidocétose diabétique ont été observés exclusivement chez les patients diabétiques.
Liste des effets indésirables
Les effets indésirables suivants ont été identifiés dans les essais cliniques contrôlés versus placebo ainsi que dans les rapports de post-commercialisation. Aucun d'entre eux ne s'est révélé dose-dépendant. Les effets indésirables mentionnés ci-dessous sont classés par fréquence et par classe de systèmes d'organes (MedDRA). Les différentes catégories de fréquence adoptent la convention suivante: très fréquents (≥1/10), fréquents (≥1/100 à <1/10), occasionnels (≥1/1000 à <1/100), rares (≥1/10'000, <1/1000), très rares (<1/10'000).
Infections et infestations
Fréquents: vulvovaginite, balanite et infections génitales associéesa, infections des voies urinairesb.
Très rares: gangrène de Fournier (fasciite nécrosante du périnée)
Affections hématologiques et du système lymphatique
Fréquent: augmentation de l'hématocrite
Troubles du métabolisme et de la nutrition
Très fréquent: hypoglycémie (en cas d'utilisation avec SU ou insuline)
Fréquents* : déplétion volémiquec, dyslipidémie.
Occasionnel: soif.
Rare: acidocétose diabétiqued (voir «Mises en garde et précautions»)
Affections gastro-intestinales
Occasionnel: constipation.
Affections de la peau et du tissu sous-cutané
Occasionnel: hyperhidrose.
Indéterminé: éruption cutanéee
Affections musculosquelettiques et systémiques
Fréquent: dorsalgie.
Affections du rein et des voies urinaires
Fréquents: dysurie, polyurief.
Occasionnels: nycturie, augmentation de l'urée sanguine.
Affections des organes de reproduction et du sein
Occasionnel: prurit vulvovaginal.
a Vulvovaginite, balanite et infections associées incluent plusieurs termes standards prédéfinis, y compris: infection et candidose vulvovaginale, balano-posthite, balanite à Candida, infections et abcès péniens, vaginite bactérienne, abcès de la vulve.
b L'infection des voies urinaires inclut les termes standards prédéfinis, y compris: infection du tractus uro-génital, cystite, pyélonéphrite, trigonite, uréthrite, prostatite.
c La déplétion volumique inclut les termes standards prédéfinis suivants: déshydratation, hypovolémie, hypotension.
d Identifié dans la grande étude des issues CV chez les patients atteints diabète de type 2. La fréquence est basée sur le taux annuel.
e L'éruption cutanée inclut les termes standards recommandés suivants: éruption cutanée, éruption généralisée, éruption prurigineuse, éruption maculaire, éruption maculo-papuleuse, éruption pustuleuse, éruption vésiculeuse, éruption érythémateuse.
f La polyurie inclut les termes standards suivants: pollakiurie, polyurie, augmentation de la production d'urine.
Description de certains effets indésirables
Acidocétose diabétique (ACD)
Dans le cadre de l'étude DECLARE évaluant la dapagliflozine chez des patients atteints de diabète de type 2, dans laquelle 8574 patients ont reçu de la dapagliflozine 10 mg et 8569 patients un placebo pendant une durée d'exposition moyenne de 48 mois, des événements d'ACD ont été rapportés chez 27 patients du groupe dapagliflozine 10 mg et 12 patients du groupe placebo. La répartition des évènements était homogène pendant la période d'évaluation. Des 27 patients avec évènements ACD dans le groupe dapagliflozine, 22 recevaient un traitement par insuline concomitant au moment de l'évènement. Les facteurs de précipitation de l'ACD étaient ceux attendus dans une population atteinte de diabète de type 2 (voir « Mises en garde et précautions »).
Dans l'étude DAPA-HF, une acidocétose diabétique a été signalée chez 3 patients atteints de diabète de type 2 dans le groupe dapagliflozine et aucun cas n'a été signalé dans le groupe placebo.
Hypoglycémie
La fréquence de l'hypoglycémie dépendait du type de traitement de fond utilisé dans chaque étude.
Dans les études portant sur la dapagliflozine en monothérapie, en association à la metformine ou en association à la sitagliptine (avec ou sans metformine), la fréquence des épisodes d'hypoglycémie mineure était similaire (<5%) entre les groupes de traitement, y compris le placebo jusqu'à 102 semaines de traitement. Dans toutes les études, des épisodes d'hypoglycémie sévère sont survenus occasionnellement avec une incidence comparable entre les groupes traités par la dapagliflozine ou le placebo. Les études en association aux sulfonylurées et aux traitements par insuline avaient des taux plus élevés d'hypoglycémie (voir «Mises en garde et précautions»).
Dans une étude en association avec le glimépiride, des épisodes d'hypoglycémie mineure ont été rapportés, respectivement aux semaines 24 et 48, plus souvent dans le groupe de patients traités par dapagliflozine 10 mg et glimépiride (6,0% et 7,9%) et dans le groupe de patients traités par dapagliflozine 5 mg plus glimépiride (5,5% et 8,3%) que dans le groupe traité par placebo plus glimépiride (2,1% et 2,1%).
Dans une étude en association avec l'insuline, des épisodes d'hypoglycémie sévère ont été rapportés, respectivement aux semaines 24 et 104, chez 0,5% et 1,0% des patients traités par dapagliflozine 10 mg et insuline, chez 0,5% des patients traités par dapagliflozine 5 mg et insuline à la semaine 24 et chez 0,5% des patients traités par placebo et insuline aux semaines 24 et 104. Aux semaines 24 et 104, des épisodes d'hypoglycémie mineure ont été rapportés respectivement chez 40,3% et 53,1% des patients sous dapagliflozine 10 mg et de l'insuline, chez 43,4% et 52,8% des patients sous dapagliflozine 5 mg plus insuline et chez 34,0% et 41,6% des patients ayant reçu le placebo plus l'insuline.
Dans l'étude DECLARE évaluant la dapagliflozine, aucune augmentation du risque d'événements d'hypoglycémie grave n'a été observée pour le traitement par dapagliflozine comparé au placebo. Des évènements d'hypoglycémie grave ont été rapportés chez 58 patients (0,7%) du groupe dapagliflozine et 83 patients (1,0%) du groupe placebo.
Dans l'étude DAPA-HF, le nombre d'événements d'hypoglycémie grave était comparable dans les deux bras de traitement, avec à chaque fois 4 patients (0,2%). Des événements d'hypoglycémie graves sont survenus uniquement chez des patients atteints de diabète de type 2.
Vulvovaginite, balanite et infections génitales associées
Des cas de vulvovaginite, de balanite et d'infections génitales associées ont été rapportés respectivement chez 5,5% et 0,6% des patients ayant reçu de la dapagliflozine 10 mg ou le placebo. La plupart des infections étaient légères à modérées et ont rarement conduit à un arrêt du traitement par dapagliflozine, les patients ayant répondu à un premier traitement standard. Ces infections étaient plus fréquentes chez les femmes (8,4% et 1,2% pour la dapagliflozine et le placebo, respectivement). Les patients avec un antécédent étaient plus susceptibles d'avoir une infection récurrente.
Dans le pool de données comprenant 12 études à court terme contrôlées versus placebo, une vulvovaginite, une balanite et des infections génitales associées ont été rapportées chez 0,9% des patients sous placebo, chez 5,7% des patients sous dapagliflozine 5 mg et chez 4,8% des patients sous dapagliflozine 10 mg. Les patients avec un antécédent étaient plus susceptibles d'avoir une infection récurrente.
Dans l'étude DAPA HF, aucun cas d'infection génitale grave n'a été signalé dans le groupe dapagliflozine, contre un cas dans le groupe placebo. Le traitement a dû être interrompu chez sept patients (0,3%) dans le groupe dapagliflozine en raison d'un événement indésirable dû à une infection génitale, et chez aucun patient dans le groupe placebo.
Infections des voies urinaires
Les infections des voies urinaires ont été plus fréquemment rapportées chez les patients sous dapagliflozine 10 mg que chez ceux ayant reçu le placebo (4,7% versus 3,5%). La plupart des infections étaient légères à modérées et ont rarement entraîné l'arrêt du traitement par dapagliflozine. Ces infections ont été plus fréquentes chez les femmes, et les patients ayant un antécédent correspondant étaient plus susceptibles d'avoir une infection récurrente. Les cas de pyélonéphrite étaient occasionnels et survenaient à une fréquence similaire à celle observée dans le groupe témoin.
Dans le pool de données comprenant 12 études à court terme contrôlées versus placebo, des infections des voies urinaires ont été rapportées chez 3,7% des patients sous placebo, chez 5,7% des patients sous dapagliflozine 5 mg et chez 4,3% des patients sous dapagliflozine 10 mg.
Le nombre de patients atteints d'infections sévères des voies urinaires était faible dans l'étude DAPA-HF et réparti de manière équilibrée: 14 patients (0,6%) dans le groupe dapagliflozine et 17 patients (0,7%) dans le groupe placebo. Le traitement a dû être interrompu en raison d'un événement indésirable dû à une infection des voies urinaires chez 5 patients (0,2%) dans chacun des deux groupes (dapagliflozine et placebo).
Effets indésirables rénaux
L'analyse des 13 études poolées à court terme contrôlées contre placebo a montré une faible augmentation temporaire de la créatinine sérique dans le groupe dapagliflozine comparé au groupe placebo (différence moyenne par rapport à la valeur initiale à la semaine 1 et la semaine 24: 0,041 mg/dL versus 0,008 mg/dL et 0,019 mg/dL versus 0,008 mg/dL).
Paramètres biologiques
Augmentation de l'hématocrite
Dans le pool de données comprenant 13 études contrôlées versus placebo, une augmentation de l'hématocrite moyen par rapport à la valeur initiale a été observée chez les patients ayant pris Forxiga. L'augmentation a débuté à la semaine 1 et a duré jusqu'à la semaine 16, à laquelle la différence moyenne maximale a été observée par rapport à la valeur initiale. A la semaine 24, la variation moyenne de l'hématocrite par rapport à la valeur initiale était de 2,30% pour la dapagliflozine 10 mg et de -0,33% pour le placebo. A la semaine 24, les valeurs de l'hématocrite étaient >55% chez 1,3% des patients traités par dapagliflozine 10 mg et chez 0,4% des patients traités par placebo.
Taux accru de phosphate sérique inorganique
À la semaine 24, dans le pool des données, constitué de 13 études contrôlées contre placebo, un taux moyen accru de phosphate sérique par rapport à la valeur initiale (augmentation moyenne de 0,13 mg/dl versus -0,04 mg/dl) a été observé chez les patients traités par la dapagliflozine par comparaison avec les patients traités par placebo. À la semaine 24, 1,7% des patients sous Forxiga 10 mg ont présenté une hyperphosphatémie (≥5,6 mg/dl dans la catégorie d'âge 17 à 65 ans, resp. ≥5,1 mg/dl dans la catégorie d'âge ≥66 ans) versus 0,9% sous placebo.
Effet sur les lipides
À la semaine 24, dans le pool de données comprenant 13 études contrôlées versus placebo, la variation moyenne en pour cent par rapport à la valeur initiale était respectivement pour la dapagliflozine 10 mg et le placebo: cholestérol total 2,5% versus 0,0%; HDL cholestérol 6,0% versus 2,7%; LDL cholestérol 2,9% versus -1,0%; triglycérides -2,7% versus -0,7%.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
La dapagliflozine n'a pas montré de toxicité chez les sujets sains pour des doses orales uniques allant jusqu'à 500 mg (soit 50 fois la dose maximale recommandée chez l'être humain). Ces sujets ont présenté une glycosurie détectable pendant une durée dose-dépendante (au moins 5 jours pour la dose de 500 mg), sans manifestation de déshydratation, d'hypotension ou de déséquilibre électrolytique et sans effet cliniquement significatif sur l'intervalle QTc. L'incidence d'hypoglycémie était similaire au placebo. Lors des études cliniques au cours desquelles des doses quotidiennes jusqu'à 100 mg (soit 10 fois la dose maximale recommandée chez l'être humain) étaient administrées pendant 2 semaines à des sujets sains et à des patients diabétiques de type 2, l'incidence d'hypoglycémie était légèrement plus élevée que sous placebo et n'était pas dose-dépendante. Le taux d'événements indésirables, y compris déshydratation ou hypotension, était comparable au placebo, et aucune modification dose-dépendante cliniquement significative n'a été observée pour les paramètres biologiques, comprenant électrolytes sériques et biomarqueurs de la fonction rénale.
Traitement
En cas de surdosage, un traitement symptomatique adapté doit être administré en fonction de l'état clinique du patient. L'élimination de la dapagliflozine par hémodialyse n'a pas été étudiée.
Propriétés/Effets
Code ATC
A10BK01
Mécanisme d'action
La dapagliflozine est un inhibiteur sélectif et réversible du co-transporteur de sodium-glucose de type 2 (SGLT2), qui améliore le contrôle glycémique chez les patients diabétiques et présente des bénéfices cardio-rénaux.
L'inhibition du SGLT2 induite par la dapagliflozine réduit la réabsorption du glucose depuis le filtrat glomérulaire dans le tubule rénal proximal, tout en diminuant la réabsorption du sodium. Il en résulte une élimination du glucose par l'urine et une diurèse osmotique. La dapagliflozine augmente ainsi l'afflux de sodium vers le tubule distal, ce qui entraîne probablement une amplification du feedback tubulo-glomérulaire et une diminution de la pression intraglomérulaire. Parmi les effets secondaires de l'inhibition du SGLT2 par la dapagliflozine, on compte également une légère diminution de la tension artérielle, la perte de poids et l'augmentation de l'hématocrite.
La dapagliflozine améliore la glycémie à jeun et postprandiale en réduisant la réabsorption rénale du glucose et en favorisant ainsi son excrétion urinaire. Cette excrétion de glucose (effet glycosurique) est observée dès la première dose déjà, reste effective durant l'intervalle posologique de 24 heures et se maintient pendant toute la durée du traitement.
La quantité de glucose éliminée par le rein via ce mécanisme dépend de la glycémie et du TFG. Chez les sujets présentant un taux normal de glucose, la dapagliflozine n'a qu'un faible potentiel d'induction d'hypoglycémie. La dapagliflozine agit indépendamment de la sécrétion et de l'action de l'insuline.
Le SGLT2 est exprimé sélectivement dans le rein. La dapagliflozine n'inhibe pas d'autres transporteurs du glucose importants pour le transport du glucose dans les tissus périphériques et est >1400 fois plus sélective pour le SGLT2 que pour le SGLT1, le principal transporteur intestinal chargé de l'absorption du glucose.
Pharmacodynamique
Une augmentation de la quantité de glucose excrétée dans l'urine a été observée chez les sujets sains et chez ceux atteints de diabète de type 2 suite à l'administration de la dapagliflozine. Près de 70 g de glucose ont été excrétés dans l'urine chaque jour avec une dose quotidienne de 10 mg de dapagliflozine administrée pendant 12 semaines à des patients atteints de diabète de type 2. Le taux d'excrétion maximale du glucose a été observé à une dose quotidienne de 20 mg de dapagliflozine. Une glycosurie était toujours manifeste chez des patients atteints de diabète de type 2 qui avaient reçu 10 mg/jour de dapagliflozine pendant 2 ans.
L'excrétion urinaire de glucose à 24 heures à l'état d'équilibre dépendait fortement de la fonction rénale. Les patients atteints d'un diabète de type 2 et présentant une fonction rénale normale ou une insuffisance rénale légère, modérée ou sévère ont ainsi respectivement éliminé 85, 52, 18 et 11 g de glucose/jour.
Cette glycosurie induite par la dapagliflozine entraîne également une diurèse osmotique ainsi qu'une augmentation du volume urinaire chez les patients présentant un diabète de type 2. L'augmentation du volume urinaire après un traitement par dapagliflozine 10 mg pendant 12 semaines et s'élevait à environ 375 ml/jour. L'augmentation du volume urinaire était associée à une augmentation légère et transitoire de l'excrétion urinaire du sodium, elle-même non liée à une modification de la concentration de sodium sérique.
L'excrétion urinaire de l'acide urique a également augmenté de manière transitoire (pendant 3 à 7 jours) et a été accompagnée d'une diminution durable de la concentration sérique d'acide urique. À 24 semaines, la diminution de la concentration sérique d'acide urique était comprise entre -18,3 à -48,3 µmol/l (de -0,33 à -0,87 mg/dl).
Efficacité clinique
Diabète
Afin d'évaluer l'efficacité et la sécurité de Forxiga, 22 études cliniques contrôlées, randomisées, menées en double aveugle ont été effectuées sur un total de 28'000 patients atteints de diabète de type 2; plus de 15'000 patients ont été traités par la dapagliflozine dans ces études. Dans la plupart des études, la durée du traitement était de 24 semaines; lors de la période d'extension en ouvert qui a suivi, les patients ont été traités sur une période allant jusqu'à 156 semaines.
L'effet de la dapagliflozine sur les évènements cardiovasculaires chez les patients avec diabète de type 2 avec ou sans maladie cardiovasculaire manifeste a été évalué dans une importante étude des issues CV (DECLARE).
Contrôle glycémique
Monothérapie
Une étude en double aveugle contrôlée versus placebo de 24 semaines (avec une période d'extension additionnelle) a été menée afin d'évaluer la tolérance et l'efficacité de la dapagliflozine en monothérapie chez des patients atteints de diabète de type 2. Le traitement par une dose quotidienne de dapagliflozine a entraîné des diminutions statistiquement significatives de l'HbA1c par rapport au placebo (Tableau 1). Durant la période d'extension, la baisse de l'HbA1c s'est maintenue jusqu'à la semaine 102 (variation moyenne ajustée par rapport aux valeurs initiales de -0,63%, -0,77% et -0,18% pour dapagliflozine 10 mg, dapagliflozine 5 mg et le placebo, respectivement).
Tableau 1. Résultats à la semaine 24 (LOCFa) d'une étude contrôlée versus placebo avec la dapagliflozine en monothérapie
Monothérapie | |||
|---|---|---|---|
Dapagliflozine | Dapagliflozine | Placebo | |
| Nb | 70 | 64 | 75 |
HbA1c (%) | |||
| Valeur initiale moyenne | 8,01 | 7,83 | 7,79 |
| Variation par rapport à la valeur initialec | -0,89 | -0,77 | -0,23 |
Différence par rapport au placeboc (IC 95%) | -0,66* (-0,96; -0,36) | -0,54* (-0,84; -0,24) | |
Poids corporel (kg) | |||
| Valeur initiale moyenne | 94,13 | 87,17 | 88,77 |
| Variation par rapport à la valeur initialec | -3,16 | -2,83 | -2,19 |
Différence par rapport au placeboc (IC 95%) | -0,97 (-2,20; 0,25) | -0,65 (-1,90; 0,61) | |
a LOCF (last observation carried forward): dernière observation reportée pour chaque patient (avant intervention de secours, le cas échéant)
b Tous les patients randomisés ayant pris au moins une dose du médicament pendant la période de l'étude en double aveugle à court terme
c Moyenne des moindres carrés ajustée par rapport à la valeur initiale
* Valeur p <0,0001 versus placebo
Traitement associé
Lors d'une étude de non-infériorité contrôlée versus un comparateur actif sur 52 semaines (avec une période d'extension de 52 semaines), la dapagliflozine a été évaluée en association à la metformine par rapport à une sulfonylurée (glipizide) en association à la metformine chez des patients dont le contrôle glycémique est inadéquat (HbA1c >6,5% et ≤10%). Les résultats ont mis en évidence une diminution moyenne de l'HbA1c entre le début de l'étude et la semaine 52 similaire à celle observée avec le glipizide, démontrant ainsi une non-infériorité (Tableau 2). À la semaine 104, la variation moyenne ajustée par rapport à la valeur initiale de l'HbA1c était de -0,32% pour la dapagliflozine 10 mg et de -0,14% pour le glipizide. Aux semaines 52 et 104, le pourcentage de patients ayant développé au moins un événement hypoglycémique était significativement moins élevé dans le groupe traité par la dapagliflozine (3,5% et 4,3% respectivement) que dans le groupe traité par le glipizide (40,8% et 47,0% respectivement). À la semaine 104, 56,2% des patients du groupe dapagliflozine étaient encore dans l'étude contre 50,0% pour le groupe glipizide.
Tableau 2. Résultats à la semaine 52 (LOCFa) d'une étude contrôlée versus produit actif comparant la dapagliflozine au glipizide en association à la metformine
Paramètres | Dapagliflozine | Glipizide |
|---|---|---|
| Nb | 400 | 401 |
HbA1c (%) | ||
| Valeur initiale moyenne | 7,69 | 7,74 |
| Variation par rapport à la valeur initialec | -0,52 | -0,52 |
Différence par rapport au glipizide + metforminec (IC 95%) | 0,00d (-0,11; 0,11) | |
Poids corporel (kg) | ||
| Valeur initiale moyenne | 88,44 | 87,60 |
| Variation par rapport à la valeur initialec | -3,22 | 1,44 |
Différence par rapport au glipizide + metforminec (IC 95%) | -4,65* (-5,14; -4,17) | |
a LOCF: dernière observation reportée pour chaque patient
b Patients randomisés et traités, avec valeur initiale et au moins 1 mesure de l'efficacité en cours d'étude
c Moyenne des moindres carrés ajustée par rapport à la valeur initiale
d Non infériorité versus l'association glipizide + metformine
* Valeur p <0,0001
La dapagliflozine en association à la metformine, au glimépiride, à la sitagliptine (avec ou sans metformine) ou à l'insuline a entraîné des diminutions statistiquement significatives de l'HbA1c à 24 semaines par rapport aux patients recevant le placebo (p <0,0001; tableaux 3 à 6).
Les diminutions de l'HbA1c constatées à la semaine 24 se sont maintenues dans les études en association (glimépiride et insuline) sur 48 semaines (glimépiride) et jusqu'à 104 semaines (insuline). A la semaine 48, en association à la sitagliptine (avec ou sans metformine), les variations moyennes ajustées par rapport à la valeur initiale pour dapagliflozine 10 mg et le placebo étaient de -0,30% et 0,38% respectivement. Pour l'étude en association à la metformine, les diminutions de l'HbA1c se sont maintenues jusqu'à la semaine 102 (variation moyenne ajustée par rapport à la valeur initiale de -0,58%, -0,78% et 0,02% pour dapagliflozine 5 mg, dapagliflozine 10 mg et le placebo, respectivement).
À la semaine 104 pour l'étude avec l'insuline (associée ou non à un hypoglycémiant oral supplémentaire), les variations moyennes ajustées de l'Hb1Ac par rapport à la valeur initiale avaient diminué de −0,71% pour dapagliflozine 5 mg, de −0,71% pour dapagliflozine 10 mg et de -0,06% pour le placebo. Aux semaines 48 et 104, la dose d'insuline est restée stable par rapport à la valeur initiale chez les patients traités par dapagliflozine 5 mg ou 10 mg à une dose moyenne dans une fourchette de 75 à 79 UI/jour. Dans le groupe placebo, l'augmentation moyenne était de 10,5 UI/jour et 18,3 UI/jour par rapport à la valeur initiale (dose moyenne de 84 UI/jour et 92 UI/jour), aux semaines 48 et 104 respectivement. À la semaine 104, 72,4% des patients du groupe dapagliflozine étaient encore dans l'étude contre 54,8% pour le groupe glipizide.
Tableau 3. Résultats d'études contrôlées versus placebo de 24 semaines (LOCFa) portant sur la dapagliflozine en association à la metformine
Traitement adjuvant associé | |||
|---|---|---|---|
Metformine1 | |||
Dapagliflozine | Dapagliflozine | Placebo | |
| Nb | 135 | 137 | 137 |
HbA1c (%) | |||
| Valeur initiale moyenne | 7,92 | 8,17 | 8,11 |
| Variation par rapport à la valeur initialec | -0,84 | -0,70 | -0,30 |
Différence par rapport au placeboc (IC 95%) | -0,54* (-0,74; -0,34) | -0,41* (-0,61; -0,21) | |
Patients (%) atteignant une HbA1c <7% Données ajustées sur la valeur initiale | 40,6** | 37,5** | 25,9 |
Poids corporel (kg) | |||
| Valeur initiale moyenne | 86,28 | 84,73 | 87,74 |
| Variation par rapport à la valeur initialec | -2,86 | -3,04 | -0,89 |
Différence par rapport au placeboc (IC 95%) | -1,97* (-2,63; -1,31) | -2,16* (-2,81; -1,50) | |
1 Metformine ≥1500 mg/jour
a LOCF: dernière observation reportée pour chaque patient (avant intervention de secours, le cas échéant)
b Tous les patients randomisés ayant pris au moins une dose du médicament pendant la période de l'étude en double aveugle à court terme
c Moyenne des moindres carrés ajustée par rapport à la valeur initiale
* Valeur p <0,0001 versus placebo + hypoglycémiant oral
** Valeur p <0,05 versus placebo + hypoglycémiant oral
Tableau 4. Résultats d'études contrôlées versus placebo de 24 semaines (LOCFa) portant sur la dapagliflozine en association au glimépiride
Traitement adjuvant associé | |||
Sulfonylurée (glimépiride1) | |||
Dapagliflozine 10 mg | Dapagliflozine 5 mg | Placebo | |
Nb | 151 | 142 | 145 |
HbA1c (%) | |||
Valeur initiale moyenne | 8,07 | 8,12 | 8,15 |
Variation par rapport à la valeur initialec | -0,82 | -0,63 | -0,13 |
Différence par rapport au placeboc (IC 95%) | -0,68* (-0,86; -0,51) | -0,49* (-0,67; -0,32) | |
Patients (%) atteignant une HbA1c <7% Données ajustées sur la valeur initiale | 31,7* | 30,3* | 13,0 |
Poids corporel (kg) | |||
Valeur initiale moyenne | 80,56 | 81,00 | 80,94 |
Variation par rapport à la valeur initialec | -2,26 | -1,56 | -0,72 |
Différence par rapport au placeboc (IC 95%) | -1,54* (-2,17; -0,92) | -0,84* (-1,47; -0,21) | |
1 Glimépiride 4 mg/jour
a LOCF: dernière observation reportée pour chaque patient (avant intervention de secours, le cas échéant)
b Tous les patients randomisés ayant pris au moins une dose du médicament pendant la période de l'étude en double aveugle à court terme
c Moyenne des moindres carrés ajustée par rapport à la valeur initiale
* Valeur p <0,0001 versus placebo + hypoglycémiant oral
Tableau 5. Résultats d'études contrôlées versus placebo de 24 semaines (LOCFa) portant sur la dapagliflozine en association à la sitagliptine (avec ou sans metformine)
Traitement adjuvant associé | ||
Inhibiteur de la DPP-4 (sitagliptine2) ± metformine1 | ||
Dapagliflozine 10 mg | Placebo | |
Nb | 223 | 224 |
HbA1c (%) | ||
Valeur initiale moyenne | 7,90 | 7,97 |
Variation par rapport à la valeur initialec | -0,45 | 0,04 |
Différence par rapport au placeboc (IC 95%) | -0,48* (-0,62; -0,34) | |
Poids corporel (kg) | ||
Valeur initiale moyenne | 91,02 | 89,23 |
Variation par rapport à la valeur initialec | -2,14 | -0,26 |
Différence par rapport au placeboc (IC 95%) | -1,89* (-2,37; -1,40) | |
1 Metformine ≥1500 mg/jour
2 Sitagliptine 100 mg/jour
a LOCF: dernière observation reportée pour chaque patient (avant intervention de secours, le cas échéant)
b Tous les patients randomisés ayant pris au moins une dose du médicament pendant la période de l'étude en double aveugle à court terme
c Moyenne des moindres carrés ajustée par rapport à la valeur initiale
* Valeur p <0,0001 versus placebo + hypoglycémiant oral
Tableau 6. Résultats à la semaine 24 (LOCFa) d'une étude contrôlée versus placebo portant sur la dapagliflozine associée à l'insuline (seule ou avec un hypoglycémiant oral)
Paramètres | Dapagliflozine 10 mg | Dapagliflozine 5 mg | Placebo | |
Nb | 194 | 211 | 193 | |
HbA1c (%) | ||||
Valeur initiale moyenne | 8,58 | 8,61 | 8,46 | |
Variation par rapport à la valeur initialec | -0,90 | -0,82 | -0,30 | |
Différence par rapport au placeboc (IC 95%) | -0,60* (-0,74; -0,45) | -0,52* (-0,66; -0,38) | ||
Poids corporel (kg) | ||||
Valeur initiale moyenne | 94,63 | 93,20 | 94,21 | |
Variation par rapport à la valeur initialec | -1,67 | -0,98 | 0,02 | |
Différence par rapport au placeboc (IC 95%) | -1,68* (-2,19; -1,18) | -1,00* (-1,50; -0,50) | ||
Dose quotidienne moyenne d'insuline (UI)1 | ||||
Valeur initiale moyenne | 77,96 | 77,10 | 73,96 | |
Variation par rapport à la valeur initialec | -1,16 | -0,61 | 5,08 | |
Différence par rapport au placeboc (IC 95%) | -6,23* (-8,84; -3,63) | -5,69* (-8,25; -3,13) | ||
a LOCF: dernière observation reportée pour chaque patient (avant ou le jour de la première augmentation de la dose d'insuline, le cas échéant)
b Tous les patients randomisés ayant pris au moins une dose du médicament pendant la période de l'étude en double aveugle à court terme
c Moyenne des moindres carrés ajustée sur la valeur initiale et présence d'un hypoglycémiant oral
* Valeur p <0,0001 versus placebo + insuline ± hypoglycémiant oral
1 Augmentation de la dose d'insuline (dont insuline à action rapide, à action intermédiaire et basale) autorisée uniquement chez les patients répondant à des critères de glycémie à jeun (GAJ) prédéfinis
2 Cinquante pour cent des patients étaient traités par de l'insuline en monothérapie au début de l'étude; 50% prenaient 1 ou 2 hypoglycémiants oraux en plus de l'insuline: dans ce dernier groupe, 80% étaient sous metformine seule, 12% recevaient une thérapie associant de la metformine et une sulfonylurée, et le reste des patients était traité par d'autres hypoglycémiants oraux.
Association de la dapagliflozine à un agoniste du récepteur du GLP-1 (GLP-1-RA).
Les effets hypoglycémiants de l'association de dapagliflozine (dose quotidienne de 10 mg) à un GLP-1-RA (exénatide à libération prolongée à une dose hebdomadaire de 2 mg) et des monothérapies avec le GLP-1-RA ou la dapagliflozine ont été évalués dans le cadre d'une étude en double aveugle, à 3 bras randomisée (1:1:1), de 28 semaines. 694 patients adultes atteints de diabète de type 2 dont le contrôle glycémique était insuffisant (HbA1c ≥8,0 et ≤12,0%) sous traitement antérieur par la metformine en monothérapie (≥1500 mg/jour; traitement par la metformine poursuivi sans changement pendant la phase de traitement) ont été inclus dans l'étude. Le critère d'évaluation principal était la variation de l'HbA1c entre le début de l'étude et la semaine 28. L'association de la dapagliflozine et du GLP-1-RA était supérieure aux deux monothérapies concernant la baisse de l'HbA1c (voir tableau 7).
Tableau 7: Effets hypoglycémiants de la dapagliflozine en association à l'exénatide à libération prolongée, de la dapagliflozine et de l'exénatide à libération prolongée chez des patients traités par la metformine (phase de traitement de 28 semaines)
Paramètres | Dapagliflozine 10 mg QD + Exénatide à libération prolongée 2 mg QW | Dapagliflozine 10 mg QD + placebo QW | Exénatide à libération prolongée 2 mg QW + placebo QD |
Nc | 228 | 230 | 227 |
HbA1c (%) | |||
Valeur initiale moyenne | 9,29 | 9,25 | 9,26 |
Variation par rapport à la valeur initialea | -1,98 | -1,39 | -1,60 |
Différence par rapport au placebo (IC 95%) | -0,59* (-0,84, -0,34) | ||
Différence par rapport à l'exénatide à libération prolongée QW (IC 95%) | -0,38§ (-0,63, -0,13) | ||
Proportion de patients ayant atteint une HbA1c <7,0%b | 44,7% | 19,1% | 26,9% |
QD = dose quotidienne, QW = dose hebdomadaire, N = Nombre de patients dans le groupe de traitement, IC= intervalle de confiance.
a La modélisation des moyennes des moindres carrés (moyennes des MC) ajustées et de la(des) différence(s) entre les différents groupes de traitement de la variation à la semaine 28 par rapport à la valeur initiale a été effectuée au moyen d'un modèle mixte à mesures répétées (MMRM; mixed model with repeated measures) en tenant compte des facteurs fixes (traitement, région, strate de valeur initiale de l'HbA1c (<9,0% ou ≥9,0%), semaine et interaction traitement-semaine) et de la covariable (valeur initiale).
b Catégories déterminées à partir des mesures continues. Imputation des non-répondeurs pour tous les patients dont les données des critères d'évaluation sont manquantes. Comparaison du traitement au moyen du test de Cochran-Mantel-Haenszel (CMH), stratification selon la valeur initiale de l'HbA1c (<9,0% oder ≥9,0%). Valeurs p selon la statistique d'association générale.
c Patients qui ont reçu au moins une fois le médicament de l'étude et pour lesquels au moins une mesure de l'HbA1c post-valeur initiale est disponible
* p <0,001.
§ p <0,01.
Toutes les valeurs p sont ajustées pour la multiplicité.
Les valeurs après l'intervention de secours et après l'arrêt prématuré du médicament de l'étude sont exclues de l'analyse.
Traitement associé initial avec la metformine
L'efficacité et la sécurité d'un traitement par la metformine XR, la dapagliflozine, la metformine XR plus dapagliflozine ont été évaluées dans le cadre de deux études contrôlées à 3 bras de 24 semaines chez au total 1236 patients naïfs de traitement atteints de diabète de type 2 insuffisamment contrôlé (HbA1c ≥7,5% et ≤12%). La dose de metformine XR était de 2000 mg dans les deux études. La dose de dapagliflozine était respectivement de 5 mg et de 10 mg dans chacune des études. Une diminution significative de l'HbA1c a été observée dans les trois bras de traitement des deux études. Le traitement associé (metformine XR plus dapagliflozine) était supérieur aux monothérapies par la metformine XR ou la dapagliflozine dans les deux études (Tableau 8).
Tableau 8: résultats de deux études contrôlées évaluant la dapagliflozine dans le cadre d'un traitement associé initial avec la metformine XR (phase de traitement de 24 semaines)
Paramètres | Dapagliflozine + | Dapagliflozine + | Metformine XR + | |||
Posologie de dapagliflozine dans l'étude | 5 mg | 10 mg | 5 mg | 10 mg | 5 mg | 10 mg |
N† | 194 | 211 | 203 | 219 | 201 | 208 |
HbA1c (%) | ||||||
Valeur initiale moyenne | 9,21 | 9,10 | 9,14 | 9,03 | 9,14 | 9,03 |
Variation par rapport à la valeur initiale (moyenne ajustée‡) | -2,05 | -1,98 | -1,19 | -1,45 | -1,35 | -1,44 |
Différence par rapport à la dapagliflozine (moyenne ajustée‡) (IC 95%) | -0,86§ (-1,11,-0,62) | -0,53§ (-0,74, -0,32) | ||||
Différence par rapport à la metformine XR (moyenne ajustée‡) (IC 95%) | -0,70§ (-0,94,-0,45) | -0,54§ (-0,75, -0,33) | -0,01¶ (-0,22, 0,20) | |||
Proportion de patients ayant atteint une HbA1c <7,0% ajustée pour la valeur initiale | 52,4% | 46,6%# | 22,5% | 31,7% | 34,6% | 35,2% |
Variation de l'HbA1c par rapport à la valeur initiale chez les patients ayant une valeur initiale de l'HbA1c ≥9% (moyenne ajustée‡) | -3,01 | -2,59# | -1,67 | -2,14 | -1,82 | -2,05 |
Différence par rapport à la dapagliflozine (moyenne ajustée‡) (IC 95%) | -1,34§ (-1,72, -0,96) | -0,45# (-0,81, -0,09) | ||||
Différence par rapport à la metformine XR (moyenne ajustée‡) (IC 95%) | -1,19§ (-1,57, -0,82) | -0,53 (-0,89, -0,18) | ||||
* LOCF (last observation carried forward): la dernière observation reportée pour chaque patient (avant l'éventuelle mise en place d'une intervention de secours) a été utilisée pour l'évaluation finale
† Tous les patients randomisés ayant pris au moins une dose du médicament pendant la période de l'étude en double aveugle à court terme
‡ Moyenne des moindres carrés ajustée pour la valeur initiale
§ valeur p <0,0001.
¶ Non infériorité versus metformine XR
# valeur p <0,05
Glycémie à jeun (fasting plasma glucose, FPG)
Le traitement par dapagliflozine 5 mg ou 10 mg en monothérapie ou en association à la metformine, au glimépiride ou à la sitagliptine (avec ou sans metformine) a entraîné une réduction statistiquement significative de la glycémie à jeun (-1,90 à -1,18 mmol/l) par rapport au placebo (-0,33 à 0,21 mmol/l). Cet effet a été observé à la semaine 1 du traitement et s'est maintenu dans les études prolongées jusqu'à la semaine 102.
Glycémie postprandiale
Le traitement par dapagliflozine 10 mg en association au glimépiride a entraîné une réduction statistiquement significative de la glycémie postprandiale à 2 heures à la semaine 24. Cet effet s'est maintenu jusqu'à la semaine 48.
Le traitement par dapagliflozine 10 mg en association à la sitagliptine (avec ou sans metformine) a entraîné une réduction statistiquement significative de la glycémie postprandiale à 2 heures à la semaine 24. Cet effet s'est maintenu jusqu'à la semaine 48.
Pression artérielle
Dans une analyse poolée de 13 études contrôlées versus placebo, le traitement par dapagliflozine 10 mg a entraîné une variation de la pression artérielle systolique de -3,7 mmHg et de la pression artérielle diastolique de -1,8 mmHg par rapport aux valeurs initiales, versus -0,5 mmHg pour la pression systolique et -0,5 mmHg pour la pression diastolique dans le groupe placebo à la semaine 24. Cet effet s'est maintenu jusqu'à la semaine 104. Dans le pool de données comprenant 12 études à court terme contrôlées versus placebo, le traitement par dapagliflozine 5 mg a entraîné une variation de la pression artérielle systolique de -3,5 mmHg par rapport à la valeur initiale et de -2,1 mmHg pour la pression diastolique.
Patients atteints d'insuffisance rénale
Insuffisance rénale modérée (TFGe ≥45 à <60 ml/min/1,73 m2)
Dans une étude contrôlée contre placebo, randomisée, menée en double aveugle, l'efficacité de la dapagliflozine a été examinée sur un total de 321 patients adultes atteints de diabète de type 2 et présentant un TFGe de ≥45 à <60 ml/min/1,73 m2 (c.-à-d. présentant un trouble modéré de la fonction rénale correspondant à une IRC 3A) ainsi qu'une stabilisation de la glycémie insuffisante sous le traitement actuel. Les patients ont été traités soit par 10 mg de dapagliflozine (N=159) soit par placebo (N=161). L'HbA1c des patients inclus était au début de l'étude en moyenne de 8,2%. Le critère d'évaluation principal, la variation de l'HbA1c par rapport à la valeur initiale, montrait une supériorité statistiquement significative à la semaine 24 pour 10 mg de dapagliflozine par rapport au placebo. La variation moyenne de l'HbA1c par rapport à la valeur initiale était de -0,37% sous dapagliflozine, de -0,03% sous placebo, correspondant à une différence thérapeutique de -0,34% (IC 95% -0,53, -0,15).
Le profil de sécurité de la dapagliflozine correspondait à celui des études antérieures menées dans la population totale de patients atteints de diabète de type 2. Dans le groupe dapagliflozine, le TFGe moyen a diminué au début du traitement (dapagliflozine: -3,39 ml/min/1,73 m2, placebo: -0,90 ml/min/1,73 m2) et est resté stable pendant la phase de traitement ultérieure d'une durée de 24 semaines. Trois semaines après l'arrêt de la dapagliflozine, aucune différence pertinente n'était plus constatée au niveau du TFGe entre le groupe recevant le traitement actif et le groupe placebo.
Dans une étude antérieure portant sur un total de N=252 diabétiques, en outre, des patients présentant un TFGe entre 30 et <45 ml/min/1,73 m2 avaient été également inclus (TFGe moyen dans cette étude: 45 ml/min/1,73 m2). Dans cette étude, aucune efficacité n'a pu être mise en évidence ni pour la dose de 5 mg ni pour la dose de 10 mg (voir «Posologie/Mode d'emploi»).
Patients avec une valeur initiale d'Hb1Ac ≥9%
Chez des patients avec une valeur initiale d'Hb1Ac ≥9,0%, le traitement en monothérapie par dapagliflozine 10 mg entraînait des baisses statistiquement significatives de l'Hb1Ac à 24 semaines tant en monothérapie (variation moyenne ajustée par rapport à la valeur initiale, respectivement: -2,04% et 0,19% pour dapagliflozine 10 mg et le placebo) qu'en traitement adjuvant par la metformine (variation moyenne ajustée par rapport à la valeur initiale, respectivement: -1,32% et -0,53% pour dapagliflozine 10 mg et le placebo).
Étude sur les issues cardiovasculaires (DECLARE)
L'effet d'un traitement par dapagliflozine sur les évènements cardiovasculaires a été analysé pendant une période d'observation médiane de 4,2 ans dans le cadre d'une étude internationale, multicentrique, en double aveugle sur les issues cardiovasculaires (DECLARE), au cours de laquelle les patients avec diabète de type 2 (durée moyenne de la maladie de 11,9 ans) ont été randomisés sur dapagliflozine (N=8582) ou placebo (N=8578) en plus de leur traitement existant. La pharmacothérapie du diabète et des autres comorbidités pouvait être ajustée pendant l'étude selon les «Standard of Care (SoC)».
Au début de l'étude, la valeur HbA1c moyenne ou de l'IMC était de 8,3% ou de 32,1 kg/m2. La population d'étude d'un âge moyen de 63,9 ans était composée à 59,4% de patients sans maladie CV établie présentant les facteurs de risque suivants: âge ≥55 (hommes) ou ≥60 ans (femmes) ainsi que dyslipidémie, hypertension ou tabagisme. Les autres 40,6% des patients présentaient déjà une maladie CV manifeste au début de l'étude. L'anamnèse indiquait une insuffisance cardiaque confirmée chez 10,0% des patients. Le TFGe moyen était de 85,2 ml/min/1,73 m2 (seuls 7,4% des patients présentaient un TFGe <60 ml/min/1,73 m2), tandis que 30,3% des patients présentaient une albuminurie.
Au début de l'étude, 82,0% des patients recevaient de la metformine pour abaisser leur glycémie ; les autres traitements antihyperglycémiants incluaient l'insuline (40,9%), les sulfonylurées (42,7%), les inhibiteurs de la DPP4 (16,8%) et les agonistes du GLP-1 (4,4%). Pour le traitement des comorbidités CV, les patients recevaient des inhibiteurs de l'ECA ou des antagonistes des récepteurs de l'angiotensine II (81,3%), des statines (75,0%), des inhibiteurs de l'agrégation plaquettaire (61,1%), de l'acide acétylsalicylique (55,5%), des bêtabloquants (52,6%), des bloquants des canaux calciques (34,9%), des diurétiques thiazidiques (22,0%) et des diurétiques de l'anse (10,5%).
Les résultats de l'analyse primaire sont résumés dans le tableau 9.
Critère d'évaluation | Dapagliflozine (N = 8582) | Placebo (N = 8578) | HR [IC à 95%] | ||
Patients avec évènement | Taux d'évènements (par 1000 patients-années) | Patients avec évènement | Taux d'évènements (par 1000 patients-années) | ||
MACE | 756 (8,8) | 22,6 | 803 (9,4) | 24,2 | 0,93 [0,84 ; 1,03] |
Décès CV | 245 (2,9) | 7,0 | 249 (2,9) | 7,1 | 0,98 [0,82 ; 1,17] |
Infarctus du myocarde | 393 (4,6) | 11,7 | 441 (5,1) | 13,2 | 0,89 [0,77 ; 1,01] |
Accident vasculaire cérébral | 235 (2,7) | 6,9 | 231 (2,7) | 6,8 | 1,01 [0,84 ; 1,21] |
Critère composite insuffisance cardiaque | 417 (4,9) | 12,2 | 496 (5,8) | 14,7 | 0,83 [0,73 ; 0,95] |
Hospitalisation pour insuffisance cardiaque | 212 (2,5) | 6,2 | 286 (3,3) | 8,5 | 0,73 [0,61 ; 0,88] |
Décès CV | 245 (2.9) | 7.0 | 249 (2.9) | 7,1 | 0,98 [0,82 ; 1,17] |
Insuffisance cardiaque
L'étude Dapagliflozin And Prevention of Adverse outcomes in Heart Failure (DAPA-HF) était une étude internationale, multicentrique, randomisée, en double aveugle, contrôlée contre placebo, réalisée auprès de 4744 patients atteints d'insuffisance cardiaque (classe fonctionnelle II-IV de la New York Heart Association [NYHA]) à fraction d'éjection réduite (fraction d'éjection du ventricule gauche [FEVG] ≤40%) visant à évaluer l'effet de la dapagliflozine, en complément d'un traitement de fond standard, sur la mortalité d'origine CV et l'aggravation de l'insuffisance cardiaque (hospitalisation pour insuffisance cardiaque et consultation d'urgence pour insuffisance cardiaque).
Les patients ayant été traités par un traitement contre l'insuffisance cardiaque correspondant au traitement standard (tableau 10) ont ont été répartis dans le bras dapagliflozine 10 mg (N=2373) ou dans le bras placebo (N=2371).
Tableau 10: Traitement concomitant de l'insuffisance cardiaque au début de l'étude
Traitement concomitant de l'insuffisance cardiaque | Pourcentage des patients au début de l'étude (%) |
Inhibiteur de l'ECA ou antagonistes des récepteurs de l'angiotensine ou inhibiteur du récepteur de l'angiotensine et de la néprilysine (ARNI) | 94% |
ARNI | 11% |
Bêtabloquants | 96% |
Antagonistes du récepteur minéralocorticoïde (ARM) | 71% |
Diurétique | 93% |
Dispositif implantable | 26% |
L'objectif principal de l'étude était d'évaluer si la dapagliflozine empêchait les décès cardiovasculaires et l'aggravation de l'insuffisance cardiaque et améliorait les symptômes de l'insuffisance cardiaque.
La période d'observation s'étendait sur une durée médiane de 18 mois. La population de patients était âgée de 66 ans en moyenne et composée à 77% d'hommes; 70% des patients étaient d'origine caucasienne, 5% d'origine africaine ou afro-américaine et 24% d'origine asiatique.
Au début de l'étude, 67,5% des patients étaient dans la classe NYHA II, 31,6% dans la classe III et 0,9% dans la classe IV. La FEVG médiane était de 32%. Dans chacun des deux groupes de traitement, 42% des patients présentaient, d'après l'anamnèse, un diabète de type 2 connu, 3% de patients supplémentaires dans chaque groupe ont également été classés comme diabétiques de type 2 suite à une valeur HbA1c ≥6,5% mesurée à l'inclusion dans l'étude et lors de la randomisation.
Les patients présentant une insuffisance rénale sévère (TFGe <30 ml/min/1,73 m2) n'étaient pas autorisés à participer à l'étude. Le TFGe moyen au début de l'étude était de 66 ml/min/1,73 m2.
Mortalité cardiovasculaire et aggravation de l'insuffisance cardiaque
La dapagliflozine 10 mg était supérieure au placebo dans la prévention des décès CV et de l'aggravation de l'insuffisance cardiaque et était associée à un effet thérapeutique uniforme sur les critères d'évaluation primaire et secondaire.
La dapagliflozine a réduit la fréquence du critère d'évaluation composite primaire, constitué des décès CV, des hospitalisations pour insuffisance cardiaque et des consultations d'urgence pour insuffisance cardiaque (HR 0,74 [IC 95%: 0,65, 0,85]; p<0,0001). Le nombre de traitements nécessaires par an (NST) était de 26 (IC 95%: 18, 46). Les courbes d'événements pour la dapagliflozine et le placebo se sont séparées à un stade précoce et ont continué à diverger pendant toute la durée de l'étude (figure 1).
Figure 1: Temps jusqu'à la première apparition du critère composite constitué des décès CV, des hospitalisations pour insuffisance cardiaque et des consultations d'urgence pour insuffisance cardiaque
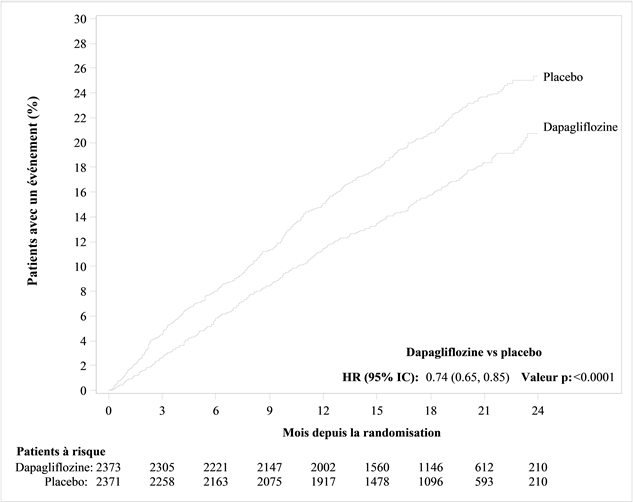
Une consultation d'urgence pour insuffisance cardiaque a été définie comme étant une évaluation urgente et non prévue effectuée par un médecin, p.ex. dans un service d'urgence, avec une nécessité thérapeutique existante (allant au-delà d'une simple augmentation de la dose de diurétiques oraux) en raison d'une aggravation de l'insuffisance cardiaque.
Les patients à risque («patients at risk») sont les patients qui étaient encore en vie au début de la période.
Les trois composants du critère d'évaluation composite primaire ont tous contribué à l'effet thérapeutique (figure 2): la dapagliflozine a réduit la fréquence des décès cardiovasculaires et des hospitalisations pour insuffisance cardiaque (HR 0,75 [IC 95%: 0,65; 0,85], p <0,0001).
Figure 2: Effets thérapeutiques pour le critère d'évaluation composite primaire, ses composants et la mortalité globale
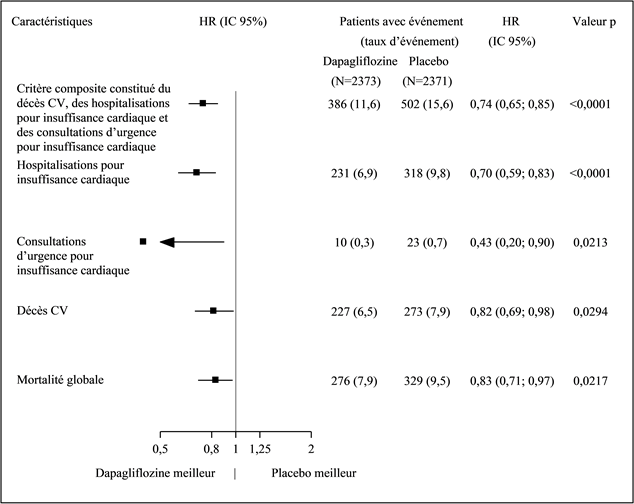
Une consultation d'urgence pour insuffisance cardiaque a été définie comme étant une évaluation urgente et non prévue effectuée par un médecin, p.ex. dans un service d'urgence, avec une nécessité thérapeutique existante (allant au-delà d'une simple augmentation de la dose de diurétiques oraux) en raison d'une aggravation de l'insuffisance cardiaque.
Le nombre de premiers événements pour chaque composant représente le nombre réel de premiers événements pour chaque composant, et leur somme n'est pas équivalente au nombre d'événements dans le critère d'évaluation composite.
Le taux d'événements est représenté comme le nombre de patients concerné par l'événement pour 100 patients-années de la période d'observation.
Les valeurs p pour les différents composants et la mortalité globale sont nominales.
Les bénéfices du traitement par dapagliflozine ont été observés chez les patients atteints d'insuffisance cardiaque, qu'ils soient atteints ou non d'un diabète de type 2 (figure 3).
Figure 3: Effets thérapeutiques chez tous les patients, c.-à-d. chez les patients avec et sans diabète de type 2
Une consultation d'urgence pour insuffisance cardiaque a été définie comme étant une évaluation urgente et non prévue effectuée par un médecin, p.ex. dans un service d'urgence, avec une nécessité thérapeutique existante (allant au-delà d'une simple augmentation de la dose de diurétiques oraux) en raison d'une aggravation de l'insuffisance cardiaque.
Le taux d'événements est représenté comme le nombre de patients concerné par l'événement pour 100 patients-années de la période d'observation.
Les bénéfices du traitement par dapagliflozine par rapport au placebo en ce qui concerne le critère d'évaluation primaire sont également cohérents dans d'autres sous-groupes importants.
Pharmacocinétique
Absorption
La dapagliflozine était bien et rapidement absorbée après administration orale. Les concentrations plasmatiques maximales de dapagliflozine (Cmax) sont généralement atteintes dans les 2 heures suivant la prise à jeun. Les moyennes géométriques des valeurs Cmax et ASCτ à l'état d'équilibre avec une dose quotidienne de 10 mg de dapagliflozine étaient respectivement de 158 ng/ml et 628 ng h/ml. La biodisponibilité orale absolue de la dapagliflozine après administration d'une dose de 10 mg est de 78%. L'administration avec un repas riche en graisses réduisait la valeur Cmax de la dapagliflozine jusqu'à 50% et allongeait la valeur Tmax d'environ 1 heure, sans toutefois modifier l'ASC par rapport à la prise à jeun. Ces changements ne sont pas considérés comme cliniquement significatifs.
Distribution
La dapagliflozine est liée à environ 91% aux protéines. La liaison protéique n'a pas été modifiée par la présence de diverses pathologies (p.ex. insuffisance rénale ou hépatique). Le volume moyen de distribution de la dapagliflozine à l'état d'équilibre était de 118 l.
Métabolisme
La dapagliflozine est largement métabolisée, principalement sous forme de 3-O-glucuronide de dapagliflozine, un métabolite inactif. Le 3-O-glucuronide de dapagliflozine ou les autres métabolites ne contribuent pas l'effet hypoglycémiant. La formation du 3-O-glucuronide de dapagliflozine est médiée par l'UGT1A9, une enzyme présente dans le foie et les reins. Le métabolisme médié par le CYP était considéré comme une voie de dégradation mineure chez l'être humain.
Élimination
La demi-vie plasmatique terminale moyenne (t1/2) de la dapagliflozine est de 12,9 heures après la prise par voie orale d'une dose unique de 10 mg de dapagliflozine chez les sujets sains. La dapagliflozine et les métabolites associés sont principalement éliminés par excrétion urinaire, avec moins de 2% de dapagliflozine sous sa forme inchangée. Après administration d'une dose de 50 mg de [14C]-dapagliflozine, 96% ont été retrouvés, 75% dans l'urine et 21% dans les selles. Dans les selles, 15% environ de la dose sont éliminés sous forme inchangée.
Linéarité
L'exposition à la dapagliflozine augmente proportionnellement à la dose dans une fourchette de 0,1 à 500 mg. Des administrations quotidiennes répétées jusqu'à 24 semaines n'ont pas modifié sa pharmacocinétique dans le temps.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Chez les patients atteints d'insuffisance hépatique légère ou modérée (classe Child-Pugh A et B), les valeurs Cmax et ASC moyennes de la dapagliflozine étaient supérieures de 12% et 36%, respectivement, à celles des témoins appariés sains. Ces différences n'ont pas été considérées comme cliniquement significatives. Chez les patients atteints d'insuffisance hépatique sévère (classe Child-Pugh C), les valeurs Cmax et ASC moyennes de la dapagliflozine étaient supérieures de 40% et 67%, respectivement, à celles des témoins appariés sains.
Troubles de la fonction rénale
À l'état d'équilibre (dose quotidienne unique de 20 mg de dapagliflozine pendant 7 jours), les patients atteints d'un diabète de type 2 et d'une insuffisance rénale légère, modérée ou sévère (déterminée par la clairance plasmatique du iohexol) présentaient une exposition systémique moyenne à la dapagliflozine supérieure de 32%, 60% et 87%, respectivement, à celle des diabétiques de type 2 ayant une fonction rénale normale. L'impact de l'hémodialyse sur l'exposition à la dapagliflozine n'est pas connu.
Patients âgés
Aucune augmentation cliniquement significative de l'exposition en fonction de l'âge seul n'a été mise en évidence chez les patients jusqu'à 70 ans. Toutefois, une exposition accrue due à la détérioration de la fonction rénale liée à l'âge peut être attendue. Il n'existe pas de données suffisantes concernant l'exposition des patients de plus de 70 ans.
Enfants et adolescents
La pharmacocinétique n'a pas été étudiée chez les enfants et les adolescents.
Sexe
Dans un modèle pharmacocinétique de population se basant sur des données issues d'études réalisées avec des sujets sains et des patients diabétiques, l'ASCee moyenne de la dapagliflozine chez les femmes (N=619) est estimée supérieure de 22% environ à celle des hommes (N=634; CI 90%: 117%, 124%).
Poids corporel
L'exposition à la dapagliflozine diminue avec l'augmentation du poids. En conséquence, les patients de faible poids peuvent avoir une exposition légèrement augmentée et les patients avec un poids élevé peuvent avoir une exposition légèrement diminuée. Toutefois, les différences d'exposition n'ont pas été considérées comme cliniquement significatives.
Origine ethnique
Aucune différence cliniquement significative n'a été observée entre les patients d'origine caucasienne, d'origine afro-américaine et d'origine asiatique concernant l'exposition systémique.
Données précliniques
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, de toxicologie en administration répétée, de génotoxicité, de potentiel carcinogène et de fertilité n'ont révélé aucun risque particulier pour l'être humain.
Mutagénicité
Pour la dapagliflozine, les résultats étaient négatifs dans les tests de mutagénicité d'Ames et positifs dans une série de tests in vitro de clastogénicité en présence d'activation métabolique (S9 mix) et à des concentrations ≥100 µg/ml. S'agissant de la clastogénicité, la dapagliflozine a donné des résultats négatifs dans une série d'études in vivo, dans lesquelles la réparation du micronoyau ou de l'ADN chez les rats était évaluée lors d'expositions >2100 fois supérieures à la dose clinique.
Carcinogénicité
La dapagliflozine n'a pas induit de tumeurs chez les souris ou les rats à chacune des doses évaluées lors des études de carcinogénicité d'une durée de deux ans. Dans une étude de 6 mois réalisée chez des rats ayant été prétraités avec 100 mg/kg de N-butyle-N-(4-hydroxybutyle)-nitrosamine pendant 6 semaines, une augmentation des hyperplasies urothéliales a été observée à la dose de 0,5 mg/kg de dapagliflozine. Aucune sur-représentation de lésions malignes n'a été enregistrée chez les animaux traités par dapagliflozine, par rapport aux animaux du groupe témoin.
Toxicité sur la reproduction
L'administration directe de dapagliflozine à de jeunes rats sevrés et l'exposition indirecte en fin de gestation (périodes correspondant aux deuxième et troisième trimestres de grossesse en matière de maturation rénale humaine) et pendant l'allaitement sont chacune associées à une incidence et/ou une sévérité accrue des dilatations du bassinet et des tubules rénaux chez la descendance.
Ces périodes d'exposition coïncident avec la phase critique de la maturation rénale chez les rats. La maturation fonctionnelle du rein se poursuivant chez l'être humain durant les 2 premières années de vie, les dilatations du bassinet et des tubules rénaux associées à la dapagliflozine et observées chez les jeunes rats pourraient représenter un risque potentiel pour la maturation du rein chez l'être humain au cours des 2 premières années de vie. De plus, les effets défavorables observés sur la prise de poids des jeunes rats sevrés et ayant été exposés pendant l'allaitement sont autant d'arguments parlant en défaveur de l'utilisation de Forxiga durant les 2 premières années de vie.
Dans une étude de toxicité juvénile, de jeunes rats ont reçu de la dapagliflozine en administration directe entre le jour postnatal 21 et le jour postnatal 90. Des dilatations du bassinet et des tubules rénaux ont été rapportées pour toutes les doses. L'exposition des petits à la plus faible dose testée était ≥15 fois supérieure à la dose maximale recommandée chez l'être humain. Ces résultats étaient associés à une augmentation dose-dépendante du poids des reins et à une hypertrophie macroscopique des reins observée à toutes les doses. Les dilatations rénales pelviennes et tubulaires constatées chez les jeunes animaux n'ont pas été totalement réversibles au cours de la période de récupération d'environ 1 mois.
Lors d'une étude distincte du développement pré- et postnatal, des rates ont été traitées à compter du jour 6 de gestation jusqu'au jour postnatal 21. Il en résulte que les petits ont été exposés indirectement in utero et durant l'allaitement. (Une étude satellite a été menée pour évaluer l'exposition à la dapagliflozine dans le lait et chez les petits). Une incidence ou une sévérité accrue des dilatations rénales pelviennes chez les descendants adultes des mères traitées a été observée, mais uniquement à la plus forte dose testée (les expositions correspondantes des mères et des petits à la dapagliflozine étaient, respectivement, 1415 fois et 137 fois supérieures aux valeurs observées à la dose maximale recommandée chez l'être humain). La toxicité additionnelle sur le développement était limitée à une perte de poids dose-dépendante chez les petits et n'a été observée qu'à des doses ≥15 mg/kg/jour (associée à une exposition ≥29 fois supérieure aux valeurs observées à la dose maximale recommandée chez l'être humain). La toxicité maternelle était uniquement évidente à la plus forte dose testée et se limitait à une perte de poids et une baisse de la prise de nourriture temporaires. La dose sans effet nocif observé (no observed adverse effect level, NOAEL) concernant la toxicité sur le développement, autrement dit la plus faible dose testée, était associée à une exposition maternelle systémique environ 19 fois supérieure aux valeurs observées à la dose maximale recommandée chez l'être humain.
Lors d'études complémentaires portant sur le développement embryo-fœtal chez les rats et les lapins, la dapagliflozine a été administrée selon des intervalles coïncidant avec les phases principales d'organogénèse de chacune des espèces. Aucune des doses testées n'a induit de toxicité maternelle ou sur le développement chez les lapins. La plus forte dose testée était associée à une exposition systémique environ 1191 fois supérieure à la dose maximale recommandée chez l'être humain. Chez les rats, la dapagliflozine ne s'est révélée ni embryolétale ni tératogène à des expositions jusqu'à 1441 fois supérieures à la dose maximale recommandée chez l'être humain.
Fertilité
Aucune des doses testées de dapagliflozine n'a entraîné d'effet sur la fertilité des rats mâles et femelles.
Remarques particulières
Influence sur les méthodes de diagnostic
Interférences entre le médicament et le test 1,5-anhydroglucitol
Il n'est pas conseillé d'utiliser le test 1,5-AG pour surveiller la glycémie des patients traités par des inhibiteurs de SGLT2, car ce traitement peut rendre les mesures du 1,5-AG non fiables pour l'évaluation du contrôle glycémique. Il faut utiliser des méthodes alternatives pour le suivi du contrôle glycémique.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 30 °C.
Tenir hors de portée des enfants
Numéro d’autorisation
65176 (Swissmedic).
Titulaire de l’autorisation
AstraZeneca AG, 6340 Baar.
Mise à jour de l’information
Juillet 2020.
Reviews (0)

Free consultation with an experienced pharmacist
Describe the symptoms or the right drug - we will help you choose its dosage or analogue, place an order with home delivery or just consult.
We are 14 pharmacists and 0 bots. We will always be in touch with you and will be able to communicate at any time.
Bestsellers

Milupa Aptamil Pepti Syneo Can 400g
Product code: 7748334Aptamil Pepti Syneo is a special food for babies from birth who are allergic to cow's milk protein a..
55.08 CHF


Weleda Baby Calendula Wind- und Wetterbalsam 30ml
Product code: 5455461Der Weleda Baby Calendula Wind- und Wetterbalsam schützt die sensible Babyhaut intensiv bei Kälte un..
15.35 CHF


HerpoTherm herpes pen
Product code: 7798882Herpotherm® - the heating pen Cold sores are not only unattractive but can also be very painful. Th..
78.48 CHF



