




Janumet XR Ret Filmtabl 50/1000 mg Fl 56 pcs
Janumet XR Ret Filmtabl 50/1000 mg Fl 56 Stk
-
84.26 CHF
- Price in reward points: 3131

- Availability: In stock
- Brand: MERCK SHARP & DOHME AG
- Product Code: 6092090
- ATC-code A10BD07
- EAN 7680650520047
Ingredients:
Ton, weisser, Metformin, Titandioxid (E171), Indigocarmin (E132), Natrium 1.78 mg, Hypromellose, Propylgallat (E310), Eisen(III)-oxid (E172), Siliciumdioxid anhydrat, Metformin hydrochlorid 1000 mg , Hyprolose, Carnaubawachs, Povidon K29-32, Sitagliptin 50 mg , Sitagliptin phosphat-1-Wasser 64.25 mg, Macrogol 3350, Natriumstearylfumarat, Überzug:.

Variants

Description
 Deutsch
Deutsch French
French Italian
ItalianWas ist Janumet XR und wann wird es angewendet?
Janumet XR ist eine Tablette, die zwei Wirkstoffe enthält, Sitagliptinphosphat und Metformin. Sitagliptin gehört zu einer Klasse von Arzneimitteln, die DPP-4 Hemmer (Dipeptidylpeptidase-4-Hemmer) genannt werden und Metformin gehört zur Klasse der Biguanide. Gemeinsam senken sie den Blutzucker von Patienten mit Diabetes mellitus Typ 2 (Zuckerkrankheit). Diabetes mellitus Typ 2 wird auch Nicht-Insulinabhängiger Diabetes mellitus genannt.
Ihr Arzt bzw. Ihre Ärztin hat Ihnen Janumet XR verschrieben, um Ihren Blutzucker zu senken, der wegen Ihrer Diabetes Typ 2 Erkrankung zu hoch ist und nicht allein durch Diät und körperliche Betätigung behandelt werden kann. Janumet XR verbessert den Insulinspiegel im Blut nach einer Mahlzeit und senkt die vom Körper produzierte Zuckermenge. Janumet XR unterstützt die Nutzung von körpereigenem Insulin.
Janumet XR darf nur auf Verschreibung des Arztes bzw. der Ärztin eingenommen werden.
Was sollte dazu beachtet werden?
Was ist Diabetes Typ 2?
Diabetes Typ 2 ist eine Krankheit, bei der Ihr Körper einerseits nicht genügend Insulin produziert und andererseits das von Ihrem Körper produzierte Insulin nicht so stark wirkt, wie es sollte. Es kann aber auch sein, dass Ihr Körper zuviel Zucker produziert. Wenn dies geschieht, reichert sich Zucker (Glukose) im Blut an. Dies kann zu gefährlichen gesundheitlichen Problemen führen.
Das Hauptziel der Behandlung von Diabetes ist die Senkung Ihres Blutzuckers auf ein normales Niveau. Die Senkung und Kontrolle des Blutzuckers kann dazu beitragen, die Komplikationen des Diabetes, wie beispielsweise Herzkrankheiten, Nierenerkrankung, Blindheit und Amputationen zu verhindern oder zu verzögern.
Hoher Blutzucker kann durch Diät und genügend Bewegung, sowie durch bestimmte Arzneimittel gesenkt werden.
Wann darf Janumet XR nicht eingenommen/angewendet werden?
Nehmen Sie Janumet XR nicht ein, wenn Sie:
- allergisch auf einen Wirkstoff oder einen Hilfsstoff dieses Arzneimittels sind
- an Diabetes Typ 1 leiden
- unter metabolischer Azidose oder diabetischer Ketoazidose leiden (erhöhte Konzentration von Ketonen im Blut oder Urin)
- schwere Nierenfunktionsprobleme haben
- Nehmen Sie Janumet XR auch nicht ein, wenn Sie Erkrankungen haben, die zu Nierenproblemen führen können wie bestimmte Herzerkrankungen, bestimmte schwerwiegende Infektionen mit. z.B. hohem Fieber, oder Durchfall und/oder Erbrechen (grosser Verlust von Körperflüssigkeiten)
- eine radiologische Untersuchung machen müssen, bei der Ihnen ein Farbstoff oder ein Kontrastmittel gespritzt wird.
Besprechen Sie mit Ihrem Arzt bzw. Ihrer Ärztin, wann Sie die Einnahme von Janumet XR beenden müssen und wann Sie es wieder einnehmen dürfen.
Wann ist bei der Einnahme/Anwendung von Janumet XR Vorsicht geboten?
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin über die folgenden aktuellen oder früheren Erkrankungen:
- Nierenprobleme.
- Leberprobleme.
- Herzprobleme, einschliesslich Herzschwäche (wenn Ihr Herz nicht genügend Blut pumpt).
- Alkoholmissbrauch (übermässiger Alkoholkonsum oder kurzfristiges Rauschtrinken).
- Wenn Sie schwanger sind oder schwanger werden wollen.
- Wenn Sie stillen.
- Wenn Sie unter Austrocknung leiden (grosser Verlust von Körperflüssigkeiten), beispielsweise bei Erkrankungen mit starkem Erbrechen, Diarrhö, Fieber oder wenn Sie viel weniger Flüssigkeit zu sich nehmen als normalerweise.
- Wenn Sie sich operieren lassen müssen.
Während der Einnahme von Janumet XR
Es wurden Fälle von Bauchspeicheldrüsenentzündungen (Pankreatitis) bei Patienten, die Janumet XR einnahmen, beobachtet. Eine Pankreatitis ist eine schwerwiegende, potentiell lebensbedrohende Krankheit. Beenden Sie die Einnahme von Janumet XR und suchen Sie Ihren Arzt bzw. Ihre Ärztin auf, falls Sie starke und anhaltende Bauchschmerzen, mit oder ohne Erbrechen bekommen, da Sie an Pankreatitis leiden könnten.
Konsultieren Sie Ihren Arzt bzw. Ihre Ärztin sofort, wenn Sie unerklärliche Muskelschmerzen, -empfindlichkeit oder -schwäche verspüren. In seltenen Fällen können Muskelprobleme schwerwiegend sein, einschliesslich Muskelabbau, der Nierenschäden zur Folge haben kann. Das Risiko kann bei Patienten mit abnormaler Nierenfunktion höher sein.
Bei Patienten, die Janumet XR erhalten, wurden Fälle einer Hautreaktion namens bullöses Pemphigoid gemeldet, die eine Behandlung in einem Spital erforderlich machen kann. Suchen Sie Ihren Arzt bzw. Ihre Ärztin auf, falls bei Ihnen Blasen oder Defekte der obersten Hautschicht (Hauterosionen) auftreten. Es kann sein, dass Ihr Arzt bzw. Ihre Ärztin entscheidet, dass Sie die Einnahme von Janumet XR beenden müssen.
Dieses Arzneimittel enthält weniger als 1 mmol (23 mg) Natrium pro Tablette, d.h. es ist im Wesentlichen «natriumfrei».
Verwendung bei Kindern und Jugendlichen
Janumet XR wurde bei Kindern und Jugendlichen unter 18 Jahren nicht untersucht und sollte von diesen nicht eingenommen werden.
Verwendung bei älteren Personen
Janumet XR sollte von älteren Patienten nur unter regelmässiger ärztlicher Kontrolle eingenommen werden.
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wenn Sie:
- an anderen Krankheiten leiden,
- Allergien haben, oder
- andere Arzneimittel (auch selbstgekaufte!) einnehmen.
Darf Janumet XR während einer Schwangerschaft oder in der Stillzeit eingenommen/angewendet werden?
Frauen, die schwanger sind oder eine Schwangerschaft planen, sollten ihren Arzt bzw. Ihre Ärztin konsultieren, bevor sie Janumet XR einnehmen. Die Einnahme von Janumet XR wird während der Schwangerschaft nicht empfohlen.
Es ist nicht bekannt, ob Janumet XR in die Muttermilch übergeht. Sie sollten deshalb Janumet XR nicht einnehmen, wenn Sie stillen oder die Absicht haben zu stillen. Sprechen Sie mit Ihrem Arzt bzw. Ihrer Ärztin.
Wie verwenden Sie Janumet XR?
Nehmen Sie Janumet XR genau so ein, wie es Ihnen von Ihrem Arzt bzw. Ihrer Ärztin verschrieben wurde. Ihr Arzt bzw. Ihre Ärztin sagt Ihnen, wie viele Tabletten Janumet XR Sie einnehmen müssen.
Es kann sein, dass Ihr Arzt bzw. Ihre Ärztin die Dosis erhöht, um eine optimale Blutzuckereinstellung zu erreichen.
Ihr Arzt bzw. Ihre Ärztin kann Ihnen Janumet XR zusammen mit bestimmten anderen Arzneimitteln verschreiben, die den Blutzucker senken, beispielsweise Sulfonylharnstoffe oder Insulin.
Nehmen Sie Janumet XR einmal pro Tag zusammen mit dem Abendessen ein, um das Risiko von Magenbeschwerden zu verringern.
Falls Sie Janumet XR einnehmen, schlucken Sie Janumet XR als ganze Tabletten herunter. Zerkauen, zerschneiden, oder zerbrechen Sie die Tabletten nicht. Informieren Sie Ihren Arzt bzw. Ihre Ärztin, falls Sie Janumet XR nicht als ganze Tabletten schlucken können.
Es kann sein, dass Sie Janumet XR Tabletten im Stuhl (Stuhlgang) entdecken. Falls Sie mehrmals Tabletten in Ihrem Stuhl sehen, sprechen Sie Ihren Arzt bzw. Ihre Ärztin darauf an. Unterbrechen Sie die Einnahme von Janumet XR nicht ohne mit Ihrem Arzt zu sprechen.
Nehmen Sie Janumet XR so lange ein, wie von Ihrem Arzt bzw. Ihrer Ärztin verschrieben, damit Ihr Blutzucker unter Kontrolle bleibt.
Es kann sein, dass Sie Janumet XR für eine kurze Zeit absetzen müssen. Informieren Sie Ihren Arzt bzw. Ihre Ärztin, wenn Sie
- unter Dehydrierung leiden (grosser Verlust von Körperflüssigkeiten), beispielsweise bei Erkrankungen mit starkem Erbrechen, Diarrhö, Fieber oder wenn Sie viel weniger Flüssigkeit zu sich nehmen als normalerweise.
- sich einer Operation unterziehen müssen.
- eine radiologische Untersuchung machen müssen, bei der Ihnen ein Farbstoff oder ein Kontrastmittel gespritzt wird.
Diät und Bewegung können dazu beitragen, dass Ihr Körper den Blutzucker besser verwertet. Es ist wichtig, dass Sie die von Ihrem Arzt bzw. Ihrer Ärztin verschriebene Diät, ausreichende Bewegung und das Programm für Gewichtsreduktion einhalten, während Sie Janumet XR einnehmen.
Falls Sie einmal eine deutlich höhere als die vorgeschriebene Dosis Janumet XR einnehmen, informieren Sie sofort Ihren Arzt bzw. Ihre Ärztin.
Sollten Sie einmal eine Dosis vergessen haben, nehmen Sie diese ein, sobald Sie sich daran erinnern. Wenn Sie sich erst daran erinnern, wenn die nächste Dosis an der Reihe ist, überspringen Sie die verpasste Dosis und fahren Sie einfach mit dem regulären Schema weiter. Nehmen Sie keine doppelte Dosis Janumet XR ein.
Ändern Sie nicht von sich aus die verschriebene Dosierung von Janumet XR. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. Ihrer Ärztin oder Apothekerin.
Welche Nebenwirkungen kann Janumet XR haben?
In seltenen Fällen kann Metformin, einer der in Janumet XR enthaltenen Wirkstoffe, eine ernsthafte Nebenwirkung verursachen, die Laktatazidose genannt wird. Laktatazidose ist ein lebensgefährlicher medizinischer Notfall, der in einem Spital behandelt werden muss. Laktatazidose wird durch eine Anreicherung von Milchsäure in Ihrem Blut verursacht.
Beenden Sie die Einnahme von Janumet XR, wenn Sie folgende Symptome einer Laktatazidose verspüren:
- Sie fühlen sich schwach und müde.
- Sie haben ungewöhnliche (anormale) Muskelschmerzen.
- Sie haben Mühe beim Atmen.
- Sie leiden unter Bauchschmerzen mit Übelkeit und Erbrechen oder Diarrhö.
- Sie verspüren Kälte, besonders in Ihren Armen und Beinen.
- Sie fühlen sich schwindlig oder leicht benommen.
- Sie haben einen langsamen oder unregelmässigen Herzschlag.
- Ihr Gesundheitszustand verändert sich plötzlich.
Die Gefahr einer Laktatazidose ist erhöht, wenn Sie:
- schwere Nierenprobleme haben.
- Leberprobleme haben.
- unter Herzschwäche leiden, die eine Behandlung mit Arzneimitteln erfordert.
- zu viel Alkohol konsumieren (sehr oft oder kurzfristiges Rauschtrinken).
- unter Austrocknung leiden (grosser Verlust von Körperflüssigkeiten). Dies kann bei Erkrankungen mit Fieber, Erbrechen oder Diarrhö der Fall sein. Eine Austrocknung kann auch auftreten, wenn Sie bei körperlicher Anstrengung viel schwitzen und dann nicht genügend Flüssigkeit zu sich nehmen.
- bestimmte radiologische Untersuchungen machen müssen, bei denen Ihnen ein Farbstoff oder ein Kontrastmittel gespritzt wird.
- bei Operationen.
- einen Herzanfall, eine schwere Infektion oder einen Schlaganfall erleiden.
Weitere häufige Nebenwirkungen von Janumet XR sind Kopfschmerzen, tiefer Blutzucker (Hypoglykämie) und Magen-Darm-Störungen, wie z.B. Durchfall, Übelkeit, Verdauungsstörungen, Blähungen, Erbrechen und metallischer Geschmack, Bauchschmerzen, Appetitverlust. In den meisten Fällen treten diese Erscheinungen zu Beginn der Behandlung auf und verschwinden dann von alleine, ohne dass die Behandlung abgebrochen werden muss. Bei längerer Behandlung mit Metformin, wird Ihr Arzt bzw. Ihre Ärztin regelmässig Ihr Blut untersuchen, da es sehr selten zu einer Verminderung von Vitamin B12 kommen kann. In vereinzelten Fällen kann es zu einer Abnahme der weissen Blutkörperchen und der Blutplättchen kommen. Ebenfalls können verstopfte oder laufende Nase, Halsentzündung, Entzündung der oberen Atemwege und Kopfschmerzen auftreten. Die Einnahme von Janumet XR zusammen mit einer Mahlzeit kann Magenproblemen vorbeugen. Wenn Sie jedoch unübliche oder unerwartete Magenprobleme haben, sollten Sie Ihren Arzt bzw. Ihre Ärztin informieren. Magenprobleme, die im späteren Verlauf der Behandlung auftreten, können ein Anzeichen für eine ernsthaftere Erkrankung sein.
Wenn Janumet XR zusammen mit einem Sulfonylharnstoff oder Insulin eingenommen wird, kann es wegen des Sulfonylharnstoffs oder Insulins zu einem tiefen Blutzucker kommen (Hypoglykämie). Ihr Arzt resp. Ihre Ärztin wird die Dosierung des Sulfonylharnstoffs reduzieren. Wenn Janumet XR in Kombination mit einem Sulfonylharnstoff eingenommen wird, kann es manchmal auch zu einer Verstopfung kommen.
Zusätzliche Nebenwirkungen können bei Einnahme von Janumet XR oder Sitagliptin, einem der Wirkstoffe von Janumet XR auftreten. Beim alleinigen Gebrauch von Janumet XR und/oder zusammen mit anderen Arzneimitteln zur Behandlung des Diabetes wurde über diese unerwünschten Wirkungen berichtet:
- Symptome einer schwerwiegenden allergischen Reaktion, können Hautausschlag, Nesselausschlag, und Schwellung von Gesicht, Lippen, Zunge und Hals, verbunden mit Atem- oder Schluckproblemen sein. Falls Sie eine allergische Reaktion haben, sollten Sie die Behandlung mit Janumet XR beenden und Ihren Arzt bzw. Ihre Ärztin sofort kontaktieren. Ihr Arzt bzw. Ihre Ärztin kann Ihnen ein Arzneimittel verschreiben, um Ihre allergische Reaktion zu behandeln und ein anderes Arzneimittel zur Behandlung ihres Diabetes.
- Es kann zu einer Entzündung der Bauchspeicheldrüse kommen. Falls Sie starke und anhaltende Bauchschmerzen, mit oder ohne Erbrechen bekommen, sollten Sie die Behandlung mit Janumet XR beenden und sofort Ihren Arzt bzw. Ärztin kontaktieren.
- Nierenprobleme (manche erfordern eine Dialyse).
- Verstopfung.
- Erbrechen.
- Gelenkschmerzen.
- Muskelschmerzen.
- Arm- oder Beinschmerzen.
- Rückenschmerzen.
- Jucken.
- Blasen auf der Haut.
Kontaktieren Sie unverzüglich Ihren Arzt bzw. Ihre Ärztin, wenn Sie Muskelschmerzen, -empfindlichkeit oder -schwäche verspüren. Dies, weil in seltenen Fällen die Muskelprobleme schwerwiegend sein können, einschliesslich Abbau von Muskelfasern, welcher zu einem Nierenschaden und im Weiteren zum Tod führen kann. Dieses Risiko ist evtl. grösser bei älteren Patienten und Patientinnen (65 Jahre und älter), beim weiblichen Geschlecht, bei Patienten und Patientinnen mit Nierenfunktionsstörungen und bei Patienten und Patientinnen mit Schilddrüsenproblemen.
Weitere Nebenwirkungen, die oben nicht aufgelistet sind, können vereinzelt bei Patienten auftreten.
Wenn Sie Nebenwirkungen bemerken, die hier nicht beschrieben sind, wenn unübliche Symptome auftauchen oder wenn bekannte Symptome anhalten oder sich verschlimmern, sollten Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin informieren.
Was ist ferner zu beachten?
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Lagerungshinweis
Bewahren Sie Janumet XR ausserhalb der Reichweite von Kindern auf.
Lagern Sie Janumet XR nicht bei Temperaturen über 30 °C.
Die Flasche ist stets gut verschlossen aufzubewahren, um die Tabletten vor Feuchtigkeit zu schützen.
Weitere Hinweise
Weitere Auskünfte erteilt Ihnen Ihr Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in Janumet XR enthalten?
Wirkstoffe
Sitagliptin als Sitagliptinphosphat-Monohydrat, Metforminhydrochlorid.
1 Tablette Janumet XR 50 mg/500 mg enthält 50 mg Sitagliptin als Sitagliptinphosphat-Monohydrat und 500 mg Metformin als Metforminhydrochlorid.
1 Tablette Janumet XR 50 mg/1000 mg enthält 50 mg Sitagliptin als Sitagliptinphosphat-Monohydrat und 1000 mg Metformin als Metforminhydrochlorid.
1 Tablette Janumet XR 100 mg/1000 mg enthält 100 mg Sitagliptin als Sitagliptinphosphat-Monohydrat und 1000 mg Metformin als Metforminhydrochlorid.
Hilfsstoffe
Propylgallat (E310), der Farbstoff Indigotin (E132), Natriumstearylfumarat, Povidon K29-32, Hydroxypropylcellulose, Hypromellose, hochdisperses Siliziumdioxid, Macrogol 3350, Kaolin, Titandioxid (E171) und Carnaubawachs.
Janumet XR 50 mg/500 mg Tabletten enthalten zudem mikrokristalline Cellulose.
Janumet XR 50 mg/1000 mg Tabletten enthalten zudem gelbes Eisenoxid (E172).
Zulassungsnummer
65052 (Swissmedic)
Wo erhalten Sie Janumet XR? Welche Packungen sind erhältlich?
In Apotheken nur gegen ärztliche Verschreibung.
Janumet XR 50 mg/500 mg ist erhältlich in Flaschen zu 56 Filmtabletten bzw. in Packungen mit 3 Flaschen zu insgesamt 168 Filmtabletten.
Janumet XR 50 mg/1000 mg ist erhältlich in Flaschen zu 56 Filmtabletten bzw. in Packungen mit 3 Flaschen zu insgesamt 168 Filmtabletten.
Janumet XR 100 mg/1000 mg ist erhältlich in Flaschen zu 28 Filmtabletten bzw. in Packungen mit 3 Flaschen zu insgesamt 84 Filmtabletten.
Zulassungsinhaberin
MSD MERCK SHARP & DOHME AG, Luzern.
Diese Packungsbeilage wurde im September 2020 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
HMV4+rhabdomyolysis/RCN000010607-CH+RCN000019721-CH
Qu'est-ce que Janumet XR et quand est-il utilisé?
Janumet XR est un comprimé qui contient deux principes actifs: le phosphate de sitagliptine et la metformine. Ces deux agents appartiennent à différentes classes de médicaments: la sitagliptine appartient à la classe des inhibiteurs de la DPP-4 (inhibiteurs de l'enzyme dipeptidyle peptidase 4), tandis que la metformine appartient à la classe des biguanides. Ensemble, les deux agents réduisent la glycémie (taux de glucose sanguin) des patients souffrant d'un diabète sucré de type 2 (maladie caractérisée par un excès de glucose sanguin). Le diabète de type 2 est aussi appelé diabète non insulinodépendant.
Votre médecin vous a prescrit Janumet XR pour réduire votre glycémie, étant donné que celle-ci est trop élevée en raison de votre diabète de type 2 et ne peut pas être suffisamment abaissée par les seuls effets d'un régime alimentaire et d'un exercice physique régulier. Janumet XR améliore le taux sanguin d'insuline après les repas et réduit la quantité de glucose produite par le corps. Janumet XR favorise l'exploitation de l'insuline fabriquée par le corps.
Janumet XR ne doit être pris que sur ordonnance du médecin.
De quoi faut-il tenir compte en dehors du traitement?
Qu'est-ce que le diabète de type 2?
Le diabète de type 2 est une maladie caractérisée par deux aspects: d'une part, votre corps ne produit plus les quantités nécessaires d'insuline; d'autre part, l'insuline produite par le corps n'agit pas aussi fortement qu'elle le devrait. Il est également possible que votre corps produise trop de glucose. Dans un tel cas, le glucose s'accumule dans le sang, ce qui peut entraîner des problèmes de santé très sérieux.
L'objectif principal du traitement du diabète est de réduire votre glycémie à un niveau normal. La réduction et le contrôle de la glycémie peuvent contribuer à empêcher ou à retarder le développement de complications du diabète, par exemple une maladie cardiaque, une maladie rénale, une cécité ou une amputation.
L'hyperglycémie (excès de glucose sanguin) peut être réduite par un régime alimentaire, un exercice physique suffisant ainsi que par certains médicaments.
Quand Janumet XR ne doit-il pas être pris/utilisé?
Ne prenez pas Janumet XR dans les cas suivants:
- si vous êtes allergique à l'un des principes actifs ou à l'un des excipients de la composition de ce médicament.
- si vous souffrez d'un diabète de type 1.
- si vous souffrez d'une acidose métabolique ou d'une acidocétose diabétique (concentration accrue de cétones dans le sang ou dans l'urine).
- si vous avez des problèmes rénaux graves.
- ne prenez pas Janumet XR si vous avez une maladie susceptible de provoquer des problèmes rénaux comme par exemple certaines maladies cardiaques, certaines infections graves accompagnées, par exemple, d'une fièvre élevée, ou des diarrhées et/ou des vomissements.
- si vous devez vous soumettre à un examen radiologique exigeant l'injection d'un colorant ou d'un agent de contraste.
Demandez à votre médecin quand vous devrez interrompre la prise de Janumet XR et quand vous pourrez à nouveau la poursuivre.
Quelles sont les précautions à observer lors de la prise/utilisation de Janumet XR?
Veuillez informer votre médecin ou votre pharmacien, si vous souffrez actuellement ou avez souffert par le passé des maladies ou conditions suivantes:
- problèmes rénaux,
- problèmes de foie,
- problèmes cardiaques, y compris insuffisance cardiaque (si votre cœur ne pompe pas suffisamment de sang),
- abus d'alcool (consommation excessive d'alcool ou consommations rapides d'alcool jusqu'à l'ivresse),
- grossesse ou désir de grossesse,
- allaitement,
- déshydratation (grandes pertes de liquides corporels), par exemple dans le cadre de vomissements importants, de diarrhée, de fièvre ou d'un apport liquidien fortement réduit par rapport à la normale,
- prévision d'une intervention chirurgicale.
Pendant la prise de Janumet XR
Des cas d'inflammation du pancréas (pancréatite) ont été observés chez des patients qui prenaient Janumet XR. La pancréatite est une maladie grave, qui peut engager le pronostic vital. Cessez de prendre Janumet XR et consultez votre médecin si vous éprouvez de fortes douleurs abdominales persistantes, avec ou sans vomissements, car il se pourrait que vous souffriez d'une pancréatite.
Veuillez consulter immédiatement votre médecin si vous ressentez des douleurs, une sensibilité ou une faiblesse au niveau des muscles qui sont inexpliquées. Dans de rares cas, les problèmes musculaires peuvent être graves, y compris une dégradation musculaire pouvant entraîner des dommages rénaux. Le risque peut être plus élevé chez les patients présentant des anomalies de la fonction rénale.
Parmi les patients qui reçoivent Janumet XR, des cas d'une réaction cutanée appelée pemphigoïde bulleuse ont été signalés. Celle-ci peut nécessiter une hospitalisation. Consultez votre médecin si des cloques ou des défauts de la couche supérieure de la peau (érosions cutanées) apparaissent. Il est possible que votre médecin décide que vous devez arrêter le traitement par Janumet XR.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d. qu'il est essentiellement «sans sodium».
Utilisation chez l'enfant et l'adolescent
Janumet XR n'a pas été étudié auprès d'enfants et d'adolescents de moins de 18 ans et ne doit pas être pris par ces personnes.
Utilisation chez les personnes âgées
Chez les patients âgés, la prise de Janumet XR exige un contrôle médical régulier.
Veuillez informer votre médecin ou votre pharmacien, si
- vous souffrez d'une autre maladie,
- vous êtes allergique, ou
- vous prenez déjà d'autres médicaments (même en automédication!).
Janumet XR peut-il être pris/utilisé pendant la grossesse ou l'allaitement?
Les femmes enceintes ou prévoyant une grossesse doivent consulter leur médecin avant de prendre Janumet XR. Une prise de Janumet XR pendant la grossesse n'est pas recommandée.
On ne sait pas si Janumet XR passe dans le lait maternel. Vous ne devez donc pas prendre Janumet XR si vous allaitez ou prévoyez de le faire. Parlez-en à votre médecin.
Comment utiliser Janumet XR?
Prenez Janumet XR exactement comme prescrit par votre médecin. Votre médecin vous dira le nombre de comprimés Janumet XR que vous devez prendre.
Il est possible que votre médecin augmente la dose pour atteindre un équilibre glycémique optimal.
Votre médecin peut vous prescrire Janumet XR en association avec certains autres médicaments hypoglycémiants tels que, par exemple, les sulfonylurées ou l'insuline.
Prenez Janumet XR une fois par jour avec le repas du soir afin de réduire le risque de problèmes gastriques.
Lorsque vous prenez Janumet XR, avalez le comprimé entier. Ne mâchez, ne coupez et ne brisez pas le comprimé. Informez votre médecin si vous n'arrivez pas à avaler le comprimé entier de Janumet XR.
Il est possible que vous trouviez des restes de comprimés de Janumet XR dans vos selles. Si vous voyez plusieurs fois des restes de comprimés dans vos selles, informez-en votre médecin. N'interrompez pas la prise de Janumet XR sans en avoir parlé avec votre médecin.
Prenez Janumet XR aussi longtemps que prescrit par votre médecin, afin que votre glycémie reste sous contrôle.
Il se peut que vous deviez suspendre la prise de Janumet XR pour une courte période. Informez votre médecin dans les cas suivants:
- si vous souffrez de déshydratation (grandes pertes de liquides corporels), par exemple dans le cadre de vomissements importants, de diarrhée, de fièvre ou d'un apport liquidien fortement réduit par rapport à la normale.
- si vous devez vous soumettre à une opération.
- si vous devez vous soumettre à un examen radiologique exigeant l'injection d'un colorant ou d'un agent de contraste.
Le régime alimentaire et l'exercice physique peuvent contribuer à une meilleure utilisation du glucose sanguin par votre corps. Il est important que vous suiviez le régime prescrit par votre médecin, fassiez suffisamment d'exercice physique et respectiez le programme de réduction pondérale pendant votre traitement par Janumet XR.
Si vous prenez une fois une dose de Janumet XR nettement supérieure à la dose prescrite, vous devez immédiatement en informer votre médecin.
Si vous oubliez une fois une dose, rattrapez-en la prise dès que vous vous en apercevez. Si vous ne vous en apercevez qu'au moment de prendre la prochaine dose, sautez la dose oubliée et poursuivez simplement votre traitement selon le schéma habituel. Ne prenez pas une double dose de Janumet XR.
Ne changez pas de votre propre chef le dosage prescrit de Janumet XR. Adressez-vous à votre médecin ou à votre pharmacien si vous estimez que l'efficacité du médicament est trop faible ou au contraire trop forte.
Quels effets secondaires Janumet XR peut-il provoquer?
Dans de rares cas, la metformine – l'un des principes actifs contenus dans Janumet XR – peut causer un effet indésirable sérieux, l'acidose lactique. L'acidose lactique est une urgence médicale susceptible d'engager le pronostic vital; elle doit être traitée à l'hôpital. L'acidose lactique est due à une accumulation d'acide lactique dans le sang.
Interrompez la prise de Janumet XR si vous ressentez les symptômes suivants d'une acidose lactique:
- vous vous sentez faible et fatigué(e).
- vous avez des douleurs musculaires anormales (inhabituelles).
- vous avez de la peine à respirer.
- vous souffrez de douleurs abdominales accompagnées de nausées, de vomissements ou de diarrhée.
- vous avez froid, en particulier aux bras et aux jambes.
- vous avez le vertige ou vous sentez légèrement engourdi(e).
- votre rythme cardiaque est lent ou irrégulier.
- votre état de santé change soudainement.
Le risque de développer une acidose lactique est accru dans les cas suivants:
- si vous avez des problèmes rénaux graves.
- si vous avez des problèmes de foie.
- si vous souffrez d'une faiblesse cardiaque exigeant un traitement médicamenteux.
- si vous consommez trop d'alcool (très souvent ou en quantité provoquant rapidement l'ivresse).
- si vous êtes déshydraté(e) (grandes pertes de liquides corporels). Ceci peut être le cas dans le cadre de maladies fébriles, de vomissements ou de diarrhées. Une déshydratation peut aussi se produire si vous transpirez fortement lors d'efforts physiques et ne buvez pas suffisamment par la suite.
- si vous devez vous soumettre à certains examens radiologiques exigeant l'injection d'un colorant ou d'un agent de contraste.
- si vous vous soumettez à une opération.
- si vous subissez une crise cardiaque, une infection sévère ou un accident vasculaire cérébral (crise d'apoplexie).
D'autres effets indésirables de Janumet XR ont englobé des maux de tête, un taux de glucose sanguin bas (hypoglycémie) et des troubles des voies digestives tels que diarrhées, nausées, problèmes de digestion, ballonnements, vomissements, goût métallique dans la bouche, mal de ventre, perte d'appétit. Dans la plupart des cas, ces manifestations apparaissent au début du traitement et disparaissent d'elles-mêmes sans exiger une interruption du traitement. Si vous devez être traité(e) pendant longtemps avec la metformine, votre médecin effectuera des analyses sanguines régulières parce qu'il est possible, dans de très rares cas, de développer un déficit en vitamine B12. Dans des cas isolés, il se peut que le nombre de globules blancs et de plaquettes sanguines soit réduit. Les effets indésirables éventuels englobent une congestion nasale (nez bouché) ou une rhinorrhée (nez qui coule), une inflammation de la gorge, une inflammation des voies aériennes supérieures et des maux de tête. La prise de Janumet XR avec un repas permet de prévenir les problèmes d'estomac. Si vous avez néanmoins des problèmes gastriques inhabituels ou inattendus, vous devez en informer votre médecin. Les problèmes gastriques apparaissant après un traitement prolongé peuvent être des indices d'une maladie plus sérieuse.
Si Janumet XR est pris en association avec une sulfonylurée ou l'insuline, celle-ci peut provoquer une hypoglycémie (glycémie trop basse). Votre médecin réduira la dose de la sulfonylurée. Lors d'une association de Janumet XR avec une sulfonylurée, on observe éventuellement parfois une constipation.
Des effets secondaires additionnels peuvent survenir lors de la prise de Janumet XR ou de sitagliptine, l'un des principes actifs de Janumet XR. Ces effets indésirables ont été rapportés lors de l'utilisation de Janumet XR, seul et/ou en association avec d'autres substances pour le traitement du diabète:
- Les symptômes d'une réaction allergique sérieuse peuvent être: éruption cutanée, urticaire, gonflement du visage, des lèvres, de la langue et du cou, en association avec des problèmes de respiration ou de déglutition. Si vous subissez une réaction allergique, vous devez interrompre la prise de Janumet XR et contacter immédiatement votre médecin. Votre médecin pourra vous prescrire un médicament pour traiter la réaction allergique et vous faire passer à un autre médicament pour le traitement de votre diabète.
- Une inflammation du pancréas est possible. Si vous éprouvez de fortes douleurs abdominales persistantes, avec ou sans vomissements, vous devez arrêter le traitement de Janumet XR et consulter immédiatement votre médecin.
- Des problèmes rénaux (certains nécessitent une dialyse).
- Constipation.
- Vomissements.
- Douleurs articulaires.
- Douleurs musculaires.
- Douleurs des bras ou des jambes.
- Douleurs dorsales.
- Démangeaison.
- Cloques sur la peau.
Veuillez immédiatement contacter votre médecin si vous présentez des douleurs, une sensibilité ou une faiblesse musculaire. En effet, dans de rares cas, les problèmes musculaires peuvent être graves et aller éventuellement jusqu'à une dégradation des fibres musculaires, pouvant entraîner des dommages rénaux et en outre la mort. Ce risque peut être plus accentué chez les patients âgés (à partir de 65 ans), chez les femmes, chez les patients souffrant d'un trouble de la fonction rénale et chez les patients présentant des problèmes de la glande thyroïde.
D'autres effets indésirables non mentionnés ci-dessus peuvent se manifester dans des cas isolés chez certains patients.
Si vous observez des effets indésirables non mentionnés ici, si vous développez des symptômes inhabituels ou constatez qu'un symptôme connu persiste ou s'aggrave, vous devriez en informer votre médecin ou votre pharmacien.
À quoi faut-il encore faire attention?
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques concernant le stockage
Tenir Janumet XR hors de la portée des enfants.
Ne pas conserver Janumet XR à des températures au-dessus de 30 °C.
Toujours conserver le flacon hermétiquement fermé à l'abri de l'humidité.
Remarques complémentaires
Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien, qui disposent d'une information détaillée destinée aux professionnels.
Que contient Janumet XR?
Principes actifs
Sitagliptine sous forme de phosphate monohydraté de sitagliptine, chlorhydrate de metformine.
1 comprimé de Janumet XR 50 mg/500 mg contient 50 mg de sitagliptine sous forme de phosphate monohydraté de sitagliptine et 500 mg de metformine sous forme de chlorhydrate de metformine.
1 comprimé de Janumet XR 50 mg/1000 mg contient 50 mg de sitagliptine sous forme de phosphate monohydraté de sitagliptine et 1000 mg de metformine sous forme de chlorhydrate de metformine.
1 comprimé de Janumet XR 100 mg/1000 mg contient 100 mg de sitagliptine sous forme de phosphate monohydraté de sitagliptine et 1000 mg de metformine sous forme de chlorhydrate de metformine.
Excipients
Gallate de propyle (E310), le colorant indigotine (E132), fumarate de stéaryl sodique, povidone K29-32, hydroxypropylcellulose, hypromellose, silice colloïdale anhydre, macrogol 3350, kaolin, dioxyde de titane (E171) et cire de carnauba.
Les comprimés Janumet XR 50 mg/500 mg contiennent également de la cellulose microcristalline.
Les comprimés Janumet XR 50 mg/1000 mg contiennent également de l'oxyde de fer jaune (E172).
Numéro d'autorisation
65052 (Swissmedic)
Où obtenez-vous Janumet XR? Quels sont les emballages à disposition sur le marché?
En pharmacie, seulement sur ordonnance médicale.
Janumet XR 50 mg/500 mg est disponible en flacons de 56 comprimés pelliculés et dans des emballages à 3 flacons de 168 comprimés pelliculés au total.
Janumet XR 50 mg/1000 mg est disponible en flacons de 56 comprimés pelliculés et dans des emballages à 3 flacons de 168 comprimés pelliculés au total.
Janumet XR 100 mg/1000 mg est disponible en flacons de 28 comprimés pelliculés et dans des emballages à 3 flacons de 84 comprimés pelliculés au total.
Titulaire de l'autorisation
MSD MERCK SHARP & DOHME AG, Lucerne.
Cette notice d'emballage a été vérifiée pour la dernière fois en septembre 2020 par l'autorité de contrôle des médicaments (Swissmedic).
HMV4+rhabdomyolysis/RCN000010607-CH+RCN000019721-CH
Che cos'è Janumet XR e quando si usa?
Janumet XR è una compressa che contiene due principi attivi: il sitagliptin fosfato e la metformina. Il sitagliptin appartiene a una classe di medicamenti denominati inibitori della DPP-4 (dipeptidil peptidasi 4), mentre la metformina fa parte della classe delle biguanidi. La loro azione combinata riduce la glicemia (livello di glucosio nel sangue) nei pazienti affetti da diabete mellito di tipo 2, detto anche diabete mellito non insulino-dipendente.
Il medico le ha prescritto Janumet XR per ridurre la glicemia che, a causa del diabete di tipo 2 di cui soffre, è troppo alta e non può essere trattata solo mediante regime dietetico e attività fisica. Janumet XR migliora il livello di insulina nel sangue dopo un pasto e riduce la quantità di glucosio prodotto dall'organismo. Janumet XR favorisce quindi l'utilizzo dell'insulina propria dell'organismo.
Janumet XR deve essere assunto solo su prescrizione medica.
Di che cosa occorre inoltre tener conto durante il trattamento?
Cos'è il diabete di tipo 2?
Il diabete di tipo 2 è una patologia in cui da un lato l'organismo non produce una sufficiente quantità di insulina e dall'altro lato l'insulina prodotta non agisce come dovrebbe. È però anche possibile che l'organismo produca un'eccessiva quantità di glucosio, che si accumula nel sangue. In questo caso si possono manifestare pericolosi problemi di salute.
Lo scopo principale del trattamento del diabete è la riduzione della glicemia a un livello normale. L'abbassamento e il controllo della glicemia possono contribuire a impedire o ritardare le complicanze del diabete quali, per esempio, malattie cardiache e renali, cecità e amputazioni.
Una glicemia elevata può essere ridotta adottando un regime dietetico specifico e praticando sufficiente attività fisica, nonché mediante assunzione di medicamenti specifici.
Quando non si può assumere/usare Janumet XR?
Non assuma Janumet XR nei seguenti casi:
- se è allergico a un principio attivo o a un eccipiente di questo medicamento;
- se è affetto da diabete di tipo 1;
- se soffre di acidosi metabolica o chetoacidosi diabetica (aumento della concentrazione di corpi chetonici nel sangue o nelle urine);
- se manifesta problemi renali gravi;
- non assuma Janumet XR anche se soffre di malattie che possono portare a problemi renali, quali alcune malattie cardiache, alcune infezioni gravi con, ad esempio, febbre alta o diarrea e/o vomito;
- se deve sottoporsi a un esame radiologico in cui viene iniettato un colorante o un mezzo di contrasto.
Chieda al suo medico quando deve interrompere l'assunzione di Janumet XR e quando la può riprendere di nuovo.
Quando è richiesta prudenza nella somministrazione/nell'uso di Janumet XR?
Informi il suo medico o il suo farmacista se soffre o ha sofferto di una delle seguenti malattie:
- problemi renali;
- problemi epatici;
- problemi cardiaci, inclusa insufficienza cardiaca (incapacità del cuore di pompare una quantità adeguata di sangue);
- abuso di alcol (consumo eccessivo di alcol regolare o consumo smodato episodico);
- se è in gravidanza o desidera iniziare una gravidanza;
- se è in allattamento;
- se soffre di disidratazione (ingente perdita di liquidi corporei), per esempio in caso di malattie che comportano vomito, diarrea o febbre grave, oppure quando assume una quantità di liquidi molto inferiore al solito;
- se deve essere sottoposto a intervento chirurgico.
Durante l'assunzione di Janumet XR
In pazienti che assumevano Janumet XR sono stati osservati casi di pancreatite (infiammazione del pancreas). La pancreatite è una malattia grave, che può condurre a morte. Se manifesta dolori addominali forti e persistenti, con o senza vomito, interrompa l'assunzione di Janumet XR e si rivolga al suo medico, poiché potrebbe soffrire di pancreatite.
Consulti immediatamente il suo medico, se dovesse avvertire dolori muscolari, sensibilità o debolezza muscolare inspiegabile. In casi rari i problemi muscolari possono essere gravi, inclusa una degenerazione muscolare, che può causare lesioni renali. Il rischio può essere più elevato nei pazienti con disfunzione renale.
In pazienti che assumono Janumet XR sono stati segnalati casi di pemfigoide bolloso, una reazione cutanea che può rendere necessario un trattamento ospedaliero. Si rivolga al suo medico se dovessero presentarsi bolle o lesioni a carico della parte più superficiale della pelle (erosioni cutanee). È possibile che il suo medico decida che si deve interrompere l'assunzione di Janumet XR.
Questo medicamento contiene meno di 1 mmol (23 mg) di sodio per compressa, cioè essenzialmente «senza sodio».
Uso nei bambini e negli adolescenti
L'impiego di Janumet XR nei bambini e negli adolescenti di età inferiore a 18 anni non è stato finora esaminato e non deve essere assunto da loro.
Uso negli anziani
Janumet XR può essere assunto da pazienti anziani solo a condizione che siano sottoposti a regolari controlli medici.
Informi il suo medico o il suo farmacista nel caso in cui:
- soffra di altre malattie,
- soffra di allergie o
- assuma altri medicamenti (anche se acquistati di sua iniziativa!).
Si può assumere/usare Janumet XR durante la gravidanza o l'allattamento?
Le donne in gravidanza o che desiderano iniziare una gravidanza devono consultare il medico prima di assumere Janumet XR. L'assunzione di Janumet XR durante la gravidanza non è raccomandata.
Dal momento che non è noto se Janumet XR passi o meno nel latte materno, non deve assumere Janumet XR se allatta o se ha intenzione di allattare. Consulti in proposito il suo medico.
Come usare Janumet XR?
Assuma Janumet XR attenendosi esattamente alle prescrizioni del suo medico. Il suo medico le dirà il numero di compresse di Janumet XR che dovrà assumere.
È possibile che il suo medico aumenti la dose per raggiungere un controllo ottimale della glicemia.
Il suo medico le può prescrivere Janumet XR in combinazione con determinati medicamenti che riducono la glicemia, come ad esempio le sulfoniluree o l'insulina.
Assuma Janumet XR una volta al giorno durante la cena per ridurre il rischio che si manifestino disturbi di stomaco.
Se prende Janumet XR, deglutisca le compresse di Janumet XR intere. Non masticare, tagliare o frantumare le compresse. Informi il suo medico se non è in grado di deglutire le compresse di Janumet XR intere.
Può succedere di ritrovare delle compresse di Janumet XR nelle feci. Se ritrova più volte delle compresse nelle feci, si rivolga al suo medico. Non interrompa l'assunzione di Janumet XR senza aver prima consultato il medico.
Affinché la sua glicemia rimanga sotto controllo, deve assumere Janumet XR per tutto il tempo prescrittole dal medico.
È possibile che debba interrompere l'assunzione di Janumet XR per un breve periodo di tempo. Informi il suo medico nei seguenti casi:
- se soffre di disidratazione (ingente perdita di liquidi corporei), per esempio in caso di malattie che comportano vomito, diarrea o febbre grave, oppure quando assume una quantità di liquidi inferiore al solito;
- se deve sottoporsi a un intervento chirurgico;
- se deve sottoporsi a un esame radiologico in cui viene iniettato un colorante o un mezzo di contrasto.
Un adeguato regime dietetico e una sufficiente attività fisica possono contribuire a migliorare l'utilizzo del glucosio da parte dell'organismo. È importante che durante l'assunzione di Janumet XR si attenga alla dieta prescrittale dal suo medico, pratichi sufficiente attività fisica e osservi il programma per la perdita di peso.
Se una volta le capitasse di assumere una dose di Janumet XR molto più elevata di quella prescritta, informi immediatamente il suo medico.
Se una volta dovesse dimenticare una dose, la prenda non appena si accorge della dimenticanza. Se si accorge della dimenticanza nel momento in cui deve assumere la dose successiva, tralasci la dose dimenticata e prosegua con lo schema posologico usuale. Non assuma una dose doppia di Janumet XR.
Non modifichi di propria iniziativa la posologia di Janumet XR prescritta. Se ritiene che l'azione del medicamento sia troppo debole o troppo forte ne parli al suo medico o al suo farmacista.
Quali effetti collaterali può avere Janumet XR?
In casi rari la metformina, uno dei principi attivi contenuti in Janumet XR, può causare un effetto collaterale grave denominato acidosi lattica. Si tratta di un'emergenza medica che può causare il decesso e che deve essere trattata in ospedale. L'acidosi lattica è causata da un accumulo di acido lattico nel sangue.
Interrompa l'assunzione di Janumet XR, se manifesta i seguenti sintomi di un'acidosi lattica:
- si sente debole e stanco;
- manifesta dolori muscolari insoliti (anomali);
- presenta una respirazione difficoltosa;
- soffre di dolori addominali accompagnati da nausea e vomito o diarrea;
- sente freddo, soprattutto alle braccia e alle gambe;
- manifesta vertigini o si sente leggermente stordito;
- ha un battito cardiaco lento o irregolare;
- il suo stato di salute cambia improvvisamente.
Il rischio di un'acidosi lattica è più elevato nei seguenti casi:
- se ha problemi renali gravi;
- se ha problemi epatici;
- se soffre di un'insufficienza cardiaca che richiede un trattamento medicamentoso;
- se assume un'eccessiva quantità di alcol (consumo molto frequente o consumo smodato episodico);
- se soffre di disidratazione (ingente perdita di liquidi corporei). Ciò può verificarsi in caso di malattie caratterizzate da febbre, vomito o diarrea. Una disidratazione può manifestarsi anche se suda molto a causa di un esercizio fisico intenso e poi non assume una sufficiente quantità di liquidi;
- se deve sottoporsi a esami radiologici specifici in cui viene iniettato un colorante o un mezzo di contrasto;
- in caso di intervento chirurgico;
- se subisce un attacco cardiaco, un'infezione grave o un ictus cerebrale.
Ulteriori effetti collaterali frequenti di Janumet XR sono mal di testa, basso livello di glucosio nel sangue (ipoglicemia) e disturbi gastrointestinali quali per es. diarrea, nausea, disturbi digestivi, meteorismo (gonfiore addominale), vomito, gusto metallico, dolori addominali e perdita d'appetito. Nella maggior parte dei casi queste manifestazioni compaiono all'inizio del trattamento e scompaiono poi da sole, senza che si debba interrompere il trattamento. In caso di trattamento prolungato con la metformina, il suo medico la sottoporrà regolarmente ad esami del sangue, poiché in casi molto rari può verificarsi una riduzione del livello di vitamina B12. In casi isolati si può manifestare un calo del numero di globuli bianchi e di piastrine. Si possono anche manifestare naso chiuso o naso che cola, infiammazione alla gola, infiammazione delle vie respiratorie superiori e mal di testa. L'assunzione di Janumet XR durante i pasti può prevenire i problemi di stomaco. Tuttavia, se presenta disturbi di stomaco insoliti o inattesi, deve informare il suo medico. I problemi di stomaco che si manifestano in fasi avanzate del trattamento possono essere il segno di una malattia più grave.
Se Janumet XR viene assunto insieme a una sulfonilurea o all'insulina, può causare una ipoglicemia (basso livello di glucosio nel sangue). Il suo medico le ridurrà la dose della sulfonilurea. Se Janumet XR viene assunto in combinazione con una sulfonilurea, in alcuni casi potrebbe causare stipsi.
Durante l'assunzione di Janumet XR o sitagliptin (uno dei principi attivi di Janumet XR) possono manifestarsi ulteriori effetti collaterali. Durante l'utilizzo di Janumet XR da solo e/o in combinazione con altri medicamenti per il trattamento del diabete sono stati segnalati questi effetti indesiderati:
- sintomi d'una reazione allergica grave possono essere eruzioni cutanee, orticaria e gonfiore al volto, alle labbra, alla lingua e alla gola, associato a difficoltà di respirazione o deglutizione. Se si dovesse manifestare una reazione allergica, deve interrompere il trattamento con Janumet XR e rivolgersi immediatamente al suo medico. Il suo medico può prescriverle un medicamento per il trattamento della reazione allergica e un altro medicamento per il trattamento del diabete;
- infiammazione del pancreas (se manifesta dolori addominali forti e persistenti, con o senza vomito, deve cessare il trattamento con Janumet XR e consultare immediatamente il medico);
- problemi renali (alcuni rendono necessaria la dialisi);
- stipsi;
- vomito;
- dolori articolari;
- dolori muscolari;
- dolori agli arti (braccia o gambe);
- mal di schiena;
- prurito;
- bolle cutanee.
Si metta immediatamente in contatto con il suo medico, se dovesse avvertire dolori muscolari, sensibilità o debolezza muscolare. Questo perché in rari casi i problemi muscolari possono essere gravi, inclusa una degenerazione delle fibre muscolari, che può causare lesioni renali ed inoltre la morte. Questo rischio può essere maggiore nei pazienti anziani (età pari o superiore a 65 anni), nei pazienti di sesso femminile, nei pazienti che soffrono di insufficienza renale e nei pazienti con problemi di tiroide.
In casi isolati i pazienti possono manifestare anche altri effetti collaterali sopra non elencati.
Se osserva effetti collaterali qui non descritti o sviluppa sintomi insoliti, oppure se sintomi noti persistono o peggiorano, dovrebbe informare il suo medico o il suo farmacista.
Di che altro occorre tener conto?
Il medicamento non deve essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Istruzioni di conservazione
Conservi Janumet XR fuori dalla portata dei bambini.
Non conservi Janumet XR a temperature superiori a 30 °C.
Il flacone va conservato sempre ben chiuso, in modo di proteggere le compresse dall'umidità.
Ulteriori indicazioni
Il medico o il farmacista, che sono in possesso di documentazione professionale dettagliata, possono darle ulteriori informazioni.
Cosa contiene Janumet XR?
Principi attivi
Sitagliptin come sitagliptin fosfato monoidrato, metformina cloridrato.
1 compressa di Janumet XR 50 mg/500 mg contiene 50 mg di sitagliptin come sitagliptin fosfato monoidrato e 500 mg di metformina come metformina cloridrato.
1 compressa di Janumet XR 50 mg/1000 mg contiene 50 mg di sitagliptin come sitagliptin fosfato monoidrato e 1000 mg di metformina come metformina cloridrato.
1 compressa di Janumet XR 100 mg/1000 mg contiene 100 mg di sitagliptin come sitagliptin fosfato monoidrato e 1000 mg di metformina come metformina cloridrato.
Sostanze ausiliarie
Propile gallato (E310), il colorante indigotina (E132), sodio stearilfumarato, povidone K29-32, idrossipropilcellulosa, ipromellosa, silice colloidale anidra, macrogol 3350, caolino, titanio diossido (E171) e cera carnauba.
Le compresse di Janumet XR 50 mg/500 mg contengono inoltre cellulosa microcristallina.
Le compresse di Janumet XR 50 mg/1000 mg contengono inoltre ferro ossido giallo (E172).
Numero dell'omologazione
65052 (Swissmedic)
Dov'è ottenibile Janumet XR? Quali confezioni sono disponibili?
In farmacia, dietro presentazione della prescrizione medica.
Janumet XR 50 mg/500 mg è disponibile in flaconi da 56 compresse rivestite con film e in confezioni con 3 flaconi da 168 compresse rivestite con film in totale.
Janumet XR 50 mg/1000 mg è disponibile in flaconi da 56 compresse rivestite con film e in confezioni con 3 flaconi da 168 compresse rivestite con film in totale.
Janumet XR 100 mg/1000 mg è disponibile in flaconi da 28 compresse rivestite con film e in confezioni con 3 flaconi da 84 compresse rivestite con film in totale.
Titolare dell'omologazione
MSD MERCK SHARP & DOHME AG, Lucerna.
Questo foglietto illustrativo è stato controllato l'ultima volta nel settembre 2020 dall'autorità competente in materia di medicamenti (Swissmedic).
HMV4+rhabdomyolysis/RCN000010607-CH+RCN000019721-CH
Zusammensetzung
Wirkstoffe
Sitagliptinphosphat-Monohydrat, Metforminhydrochlorid.
Hilfsstoffe
Povidon K29-32, Hypromellose, Hydroxypropylcellulose, hochdisperses Siliziumdioxid, Natriumstearylfumarat (jede 50 mg/500 mg Tablette enthält 1,26 mg Natrium; jede 50 mg/1000 mg Tablette enthält 1,78 mg Natrium; jede 100 mg/1000 mg Tablette enthält 1,77 mg Natrium), Propylgallat (E310), Macrogol 3350, Kaolin, Titandioxid (E171) Carnaubawachs und Indigotin (E132).
Janumet XR 50 mg/500 mg Tabletten enthalten zudem mikrokristalline Cellulose. Janumet XR 50 mg/1000 mg Tabletten enthalten zudem gelbes Eisenoxid (E172).
Darreichungsform und Wirkstoffmenge pro Einheit
Tablette mit veränderter Wirkstofffreisetzung mit 50 mg Sitagliptin als Sitagliptinphosphat-Monohydrat und 500 mg Metforminhydrochlorid mit verzögerter Wirkstofffreisetzung (Janumet XR 50 mg/500 mg), 1000 mg Metforminhydrochlorid mit verzögerter Wirkstofffreisetzung (Janumet XR 50 mg/1000 mg), sowie 100 mg Sitagliptin als Sitagliptinphosphat-Monohydrat und 1000 mg Metforminhydrochlorid mit verzögerter Wirkstofffreisetzung (Janumet XR 100 mg/1000 mg).
Indikationen/Anwendungsmöglichkeiten
Janumet XR ist als Ergänzung zu einer Diät und körperlicher Aktivität zur Einstellung des Blutzuckerspiegels bei Patienten mit Diabetes mellitus Typ 2 indiziert, wenn weder mit Metformin noch mit Sitagliptin als Monotherapie eine ausreichende Kontrolle der Glykämie erreicht wird, oder bei Patienten, die bereits eine Kombination von Sitagliptin und Metformin erhalten.
Janumet XR ist auch als Ergänzung zu einer Diät und körperlicher Aktivität in Kombination mit einem Sulfonylharnstoff (als dreifache Kombinationstherapie) bei Patienten mit Diabetes mellitus Typ 2 indiziert, wenn durch eine Kombinationstherapie von zwei beliebigen der folgenden drei Wirkstoffe keine ausreichende Kontrolle der Glykämie erreicht wird: Metformin, Sitagliptin oder ein Sulfonylharnstoff.
Wenn der Blutzuckerspiegel durch Diät und vermehrte körperliche Aktivität und Insulin alleine nicht ausreichend gesenkt werden kann: In Kombination mit Insulin.
Dosierung/Anwendung
Allgemeines
Die Dosierung der antihyperglykämischen Therapie mit Janumet XR sollte individuell auf der gegenwärtigen Therapie des Patienten, der Wirksamkeit und der Verträglichkeit basieren, wobei die empfohlene tägliche Maximaldosis von 100 mg Sitagliptin nicht überschritten werden sollte.
Janumet XR sollte einmal täglich zu einer Mahlzeit vorzugsweise am Abend eingenommen werden.
Die Dosis sollte graduell erhöht werden, um allfällige gastrointestinale Beschwerden wegen Metformin zu vermindern.
Die Tabletten dürfen vor dem Herunterschlucken nicht zerteilt, zerbrochen, zermahlen oder zerkaut werden, damit die modifizierten Freisetzungseigenschaften erhalten bleiben.
Die Plasmakonzentrationen von Metformin erhöhen sich bei Einnahme von Janumet XR zusammen mit Nahrungsmitteln. Es wurde über unvollständig aufgelöste Janumet XR Tabletten berichtet, die im Stuhl ausgeschieden wurden. Es ist nicht bekannt, ob dieses im Stuhl gefundene Material Wirkstoff enthält. Die Blutzuckereinstellung sollte daher bei Patienten, welche wiederholt über Tablettenreste im Stuhl berichten, besonders sorgfältig kontrolliert werden.
Therapieänderungen bei Diabetes mellitus Typ 2 müssen wegen der möglichen Veränderungen der glykämischen Kontrolle stets mit Vorsicht und unter angemessener Überwachung erfolgen. Dies gilt auch bei einem Wechsel von Metformin Tabletten mit nicht verzögerter Wirkstofffreisetzung auf Metformin Tabletten mit verzögerter Wirkstofffreisetzung, wie z.B. Janumet XR.
Dosierungsempfehlung
Die Initialdosis von Janumet XR sollte auf der bereits bestehenden Therapie basieren.
Janumet XR wird einmal täglich zu einer Mahlzeit vorzugsweise am Abend eingenommen. Janumet XR Tabletten sind in folgenden Dosierungen erhältlich:
50 mg Sitagliptin/500 mg Metforminhydrochlorid mit verzögerter Wirkstofffreisetzung
50 mg Sitagliptin/1000 mg Metforminhydrochlorid mit verzögerter Wirkstofffreisetzung
100 mg Sitagliptin/1000 mg Metforminhydrochlorid mit verzögerter Wirkstofffreisetzung
Patienten, die die Tablette 50 mg Sitagliptin/500 mg Metformin mit verzögerter Wirkstofffreisetzung oder die Tablette 50 mg Sitagliptin/1000 mg Metformin mit verzögerter Wirkstofffreisetzung verwenden, sollen einmal täglich zwei Tabletten zusammen einnehmen. Die Tablette 100 mg Sitagliptin/1000 mg Metformin mit verzögerter Wirkstofffreisetzung soll als einzelne Tablette einmal täglich eingenommen werden.
Patienten, bei denen mit einer Metformin-Monotherapie eine ungenügende Blutzuckereinstellung erreicht wird
Für Patienten, bei denen mit einer Metformin-Monotherapie eine ungenügende Blutzuckereinstellung erreicht wird, beträgt die empfohlene Initialdosis von Janumet XR pro Tag insgesamt 100 mg Sitagliptin und die schon vorher verschriebene Dosis Metformin.
Patienten, bei denen mit einer Sitagliptin-Monotherapie eine ungenügende Blutzuckereinstellung erreicht wird
Für Patienten, bei denen mit einer Sitagliptin-Monotherapie eine ungenügende Blutzuckereinstellung erreicht wird, beträgt die empfohlene Initialdosis von Janumet XR pro Tag insgesamt 100 mg Sitagliptin und 1000 mg Metforminhydrochlorid. Die Metformin-Dosis kann je nach Bedarf zur Einstellung des Blutzuckerspiegels titriert werden. Eine graduelle Dosissteigerung sollte in Betracht gezogen werden, um allfällige gastrointestinale Beschwerden wegen Metformin zu vermindern. Patienten mit einer Sitagliptin-Monotherapie, deren Dosis wegen einer Nierenfunktionsstörung angepasst wurde, sollten nicht auf Janumet XR umgestellt werden (siehe KONTRAINDIKATIONEN).
Patienten, die von einer Kombinationstherapie mit Sitagliptin und Metformin zu Janumet XR wechseln
Patienten, die von einer Kombinationstherapie mit Sitagliptin und Metformin zu Janumet XR wechseln, kann dieselbe Initialdosis Sitagliptin und Metformin in Form des Kombinationspräparates Janumet XR verabreicht werden wie vorher.
Patienten, bei denen mit einer Kombinationstherapie von zwei beliebigen der folgenden drei Antidiabetika keine genügende Blutzuckereinstellung möglich ist: Sitagliptin, Metformin oder ein Sulfonylharnstoff
Die übliche Initialdosis Janumet XR sollte insgesamt 100 mg Sitagliptin pro Tag enthalten. Bei der Festlegung der Initialdosis der Metformin-Komponente muss der gegenwärtige Blutzuckerspiegel und die bisherige Dosierung von Metformin, falls dieses schon vorher eingenommen wurde, berücksichtigt werden. Eine graduelle Dosissteigerung sollte vorgenommen werden, um allfällige gastrointestinale Beschwerden wegen Metformin zu vermindern. Bei Patienten, die gegenwärtig einen Sulfonylharnstoff einnehmen oder mit einer Sulfonylharnstoff-Therapie beginnen, empfiehlt es sich, die Sulfonylharnstoffdosis tiefer anzusetzen, um das Risiko einer durch Sulfonylharnstoff induzierten Hypoglykämie zu reduzieren (siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN).
Patienten, bei denen mit einer Kombinationstherapie von zwei beliebigen der folgenden drei Antidiabetika keine genügende Blutzuckereinstellung möglich ist: Sitagliptin, Metformin oder Insulin:
Die übliche Initialdosis Janumet XR sollte insgesamt 100 mg Sitagliptin pro Tag enthalten. Bei der Festlegung der Initialdosis der Metformin-Komponente muss der gegenwärtige Blutzuckerspiegel und die bisherige Dosierung von Metformin, falls dieses schon vorher eingenommen wurde, berücksichtigt werden. Eine graduelle Dosissteigerung sollte vorgenommen werden, um allfällige gastrointestinale Beschwerden wegen Metformin zu vermindern. Bei Patienten, die gegenwärtig mit Insulin behandelt werden oder mit einer Insulin-Therapie beginnen, empfiehlt es sich, die Insulindosis tiefer anzusetzen, um das Risiko einer Hypoglykämie zu reduzieren (siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN).
Es wurden keine spezifischen Studien über die Sicherheit und Wirksamkeit von Janumet XR bei Patienten durchgeführt, die vorher mit anderen oralen Antidiabetika behandelt wurden und dann auf Janumet XR umstellten. Jede Änderung der Therapie bei Diabetes mellitus Typ 2 sollte mit Vorsicht und unter genauer Kontrolle durchgeführt werden, weil Veränderungen der Blutzuckereinstellung auftreten können.
Anwendung bei Patienten mit Nierenfunktionsstörung
Die Nierenfunktion ist vor Therapiebeginn mit Janumet XR und danach in regelmässigen Abständen zu beurteilen.
Janumet XR ist kontraindiziert bei Patienten mit einer geschätzten glomerulären Filtrationsrate (eGFR) <30 ml/min/1.73 m2. Janumet XR ist abzusetzen, falls die eGFR des Patienten später unter 30 ml/min/1.73 m2 fällt (siehe KONTRAINDIKATIONEN und WARNHINWEISE UND VORSICHTSMASSNAHMEN).
Der Beginn einer Behandlung mit Janumet XR bei Patienten mit einer eGFR ≥30 ml/min/1.73 m2 und <45 ml/min/1.73 m2 ist nicht empfohlen. Bei Patienten, die Janumet XR einnehmen und deren eGFR später unter 45 ml/min/1.73 m2 fällt, ist der Nutzen und das Risiko der Therapiefortsetzung zu beurteilen und die Dosis der Sitagliptin-Komponente auf 50 mg einmal pro Tag zu limitieren.
Anwendung bei Kindern und Jugendlichen
Die Sicherheit und Wirksamkeit von Janumet XR bei Kindern und Jugendlichen wurde nicht untersucht.
Anwendung bei älteren Patienten
Da Sitagliptin und Metformin hauptsächlich durch die Niere ausgeschieden werden, sollte Janumet XR mit zunehmendem Alter des Patienten mit Vorsicht angewendet werden. Eine Kontrolle der Nierenfunktion ist besonders bei älteren Patienten zur Verhütung der metformin-assoziierten Laktatazidose notwendig (siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN, Metforminhydrochlorid, Laktatazidose).
Kontraindikationen
Janumet XR ist kontraindiziert bei Patienten mit:
- Schwerer Nierenfunktionsstörung (eGFR <30 ml/min/1.73 m2) (siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN, Metforminhydrochlorid, Nierenfunktionsstörung).
- Akuten Erkrankungen, die zu einer Beeinträchtigung der Nierenfunktion führen können, z.B. kardiovaskuläre Erkrankungen (einschliesslich Myokardinfarkt), Sepsis, Dehydrierung (Diarrhöe, wiederholtes Erbrechen), schwere Infektionen, z.B. der Harnwege, hohes Fieber.
- Bekannter Überempfindlichkeit auf Sitagliptinphosphat, Metforminhydrochlorid oder irgendeinen anderen Inhaltsstoff von Janumet XR (siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN, Überempfindlichkeitsreaktionen und UNERWÜNSCHTE WIRKUNGEN, Postmarketing Erfahrungen).
- Akuter oder chronischer metabolischer Azidose, einschliesslich diabetischer Ketoazidose mit oder ohne Koma.
- Die intravaskuläre Applikation von iodhaltigen Kontrastmitteln für Röntgenuntersuchungen kann zu einem Nierenversagen und somit zu einer Metforminakkumulation und Laktatazidose führen. Die Behandlung mit Metformin muss 48 h vor einer solchen Untersuchung unterbrochen werden, falls die Kreatinin-Clearance <60 ml/min, resp. die eGFR <60 ml/min/1.73 m2 beträgt. Die Therapie mit Metformin darf nur fortgeführt werden, wenn eine Überprüfung der Nierenfunktion 48 h nach der Kontrastmitteluntersuchung keine weitere Verschlechterung ergeben hat (siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN).
Warnhinweise und Vorsichtsmassnahmen
Janumet XR
Janumet XR sollte bei Patienten mit Diabetes Typ 1 oder für die Behandlung der diabetischen Ketoazidose nicht verwendet werden.
Pankreatitis: Es wurden bei Patienten unter Sitagliptin Fälle von akuter Pankreatitis beobachtet, inklusive tödlich und nicht tödlich verlaufene Fälle von hämorrhagischer oder nekrotisierender Pankreatitis (siehe UNERWÜNSCHTE WIRKUNGEN). Die Patienten sollten über die charakteristischen Symptome einer akuten Pankreatitis informiert werden: persistierende, schwere Abdominalschmerzen. Eine Besserung der Pankreatitis wurde nach Absetzen von Sitagliptin beobachtet. Bei Verdacht auf eine Pankreatitis sollte die Behandlung mit Janumet XR und anderen potentiell verdächtigen Arzneimitteln gestoppt werden.
Kontrolle der Nierenfunktion: Sitagliptin und Metformin werden hauptsächlich durch die Niere ausgeschieden. Vor Beginn einer Therapie mit Janumet XR soll die Nierenfunktion untersucht werden, da das Risiko einer Metforminakkumulation und Laktatazidose mit zunehmender Einschränkung der Nierenfunktion ansteigt. Janumet XR ist kontraindiziert bei Patienten mit einer schweren Nierenfunktionsstörung (eGFR <30 ml/min/1.73 m2).
Die Nierenfunktion soll unter der Behandlung mit Janumet XR in regelmässigen Abständen erneut bestimmt werden, mindestens:
- einmal jährlich bei Patienten mit normaler Nierenfunktion;
- mindestens alle 3 – 6 Monate bei Patienten mit einer eGFR von ≥45 – 59 ml/min/1.73 m2, sowie bei älteren Patienten;
- mindestens alle 3 Monate bei Patienten mit einer eGFR von ≥30 – 44 ml/min/1.73 m2.
Janumet XR soll sofort abgesetzt werden, wenn die eGFR unter 30 ml/min/1.73 m2 sinkt.
In Verbindung mit Medikamenten, welche Hypoglykämie hervorrufen können: Es ist bekannt, dass Antidiabetika vom Typ der Sulfonylharnstoffe oder Insuline Hypoglykämien verursachen können. Deshalb kann bei Anwendung dieser Substanzen in Kombination mit Janumet XR eine Reduktion der Insulin- bzw. Sulfonylharnstoff-Dosierung notwendig sein.
Sitagliptinphosphat
Myopathie/Rhabdomyolyse
Im Zusammenhang mit der Einnahme von Janumet XR wurde über Myopathien berichtet, welche sich in Form von Muskelschmerz, -schwäche oder -empfindlichkeit zusammen mit stark erhöhter Kreatinkinase (CK, auf das Zehnfache der oberen Normgrenze) äussern. Myopathie kann manchmal in Form einer Rhabdomyolyse mit oder ohne akutem Nierenversagen aufgrund einer Myoglobinurie auftreten, und selten sind Todesfälle vorgekommen.
Die Ärzte sollten Janumet XR mit Vorsicht bei Patienten mit prädisponierenden Faktoren für eine Rhabdomyolyse verschreiben. Ein Kreatinkinase-Wert sollte vor Beginn der Behandlung in den folgenden Situationen bestimmt werden:
- Einschränkung der Nierenfunktion
- Nicht-kontrollierte Hypothyreose
- Persönliche oder familiäre Vorgeschichte von erblichen Muskelerkrankungen
- Vorgeschichte von Muskeltoxizität mit einem Statin oder Fibrat
- Alkoholabhängigkeit
- Ältere Personen (≥65 Jahre): die Notwendigkeit einer solchen Messung sollte bei Vorhandensein von anderen prädisponierenden Faktoren für eine Rhabdomyolyse in Betracht gezogen werden
- Weibliches Geschlecht: die Notwendigkeit einer solchen Messung sollte bei Vorhandensein von anderen prädisponierenden Faktoren für eine Rhabdomyolyse in Betracht gezogen werden
In solchen Situationen sollte das Risiko einer Behandlung in Relation zum möglichen Nutzen betrachtet werden.
In Verbindung mit Medikamenten, welche Hypoglykämie hervorrufen können: In klinischen Studien mit Sitagliptin als Monotherapie und als Bestandteil einer Kombinationstherapie mit Wirkstoffen, die nicht mit Hypoglykämie assoziiert sind (z.B. Metformin oder PPARγ-Agonisten (Thiazolidindione)), war die Häufigkeit einer Hypoglykämie in Zusammenhang mit der Einnahme von Sitagliptin etwa gleich gross wie diejenige bei Patienten, die Placebo einnahmen.
Überempfindlichkeitsreaktionen
Es gibt Berichte nach Markteinführung über schwerwiegende Überempfindlichkeitsreaktionen bei Patienten, die mit Sitagliptin, einem der Wirkstoffe von Janumet XR, behandelt wurden. Diese Reaktionen beinhalten Anaphylaxie, Angioödem und exfoliative Hautveränderungen einschliesslich Stevens-Johnson Syndrom. Diese Reaktionen traten innerhalb der ersten 3 Monate nach Beginn der Behandlung mit Sitagliptin auf, einige sogar nach der ersten Dosis. Da diese Ereignisse aus einer Population unbekannter Grösse stammen, ist es nicht möglich, verlässliche Angaben bezüglich Häufigkeit zu machen. Falls eine Überempfindlichkeitsreaktion vermutet wird, sollte die Therapie mit Janumet XR abgesetzt werden (siehe KONTRAINDIKATIONEN und UNERWÜNSCHTE WIRKUNGEN, Postmarketing Erfahrungen).
Bullöses Pemphigoid
Nach Markteinführung wurden Fälle von bullösem Pemphigoid unter Anwendung von Dipeptidylpeptidase-4 (DPP-4) Inhibitoren gemeldet, die eine Hospitalisierung erforderten. In den gemeldeten Fällen erholten sich die Patienten typischerweise unter topischer oder systemischer immunosuppressiver Behandlung und nach Absetzen des DPP-4-Inhibitors. Patienten sind darauf hinzuweisen, bei Entwicklung von Blasen oder Erosionen der Haut unter der Behandlung mit Janumet XR ihren Arzt zu kontaktieren. Janumet XR sollte bei Verdacht auf ein bullöses Pemphigoid abgesetzt werden. Zur Sicherung der Diagnose und einer adäquaten Behandlung sollte eine Überweisung an einen Dermatologen in Betracht gezogen werden.
Metforminhydrochlorid
Laktatazidose: Laktatazidose ist eine seltene, aber schwerwiegende metabolische Komplikation, welche durch eine Akkumulation von Metformin bei der Behandlung mit Janumet XR (Sitagliptinphosphat/Metforminhydrochlorid mit verzögerter Wirkstofffreisetzung) verursacht werden kann. Die Laktatazidose verläuft in ungefähr 50% der Fälle tödlich. Laktatazidose kann auch unter verschiedenen pathophysiologischen Zuständen auftreten, einschliesslich Diabetes mellitus und immer dann, wenn eine signifikante Gewebe-Hypoperfusion und Gewebe-Hypoxie auftritt. Typisch für die Laktatazidose ist ein erhöhter Laktatspiegel im Blut (>5 mmol/l), ein erniedrigter pH des Blutes, Elektrolytstörungen mit vergrösserter Anionenlücke und ein erhöhtes Laktat/Pyruvat-Verhältnis. Wenn Metformin der Grund für die Laktatazidose ist, werden normalerweise Metformin-Plasmaspiegel von >5 µg/ml gefunden.
Die Häufigkeit der Laktatazidose ist bei Patienten, die Metforminhydrochlorid erhalten, sehr gering (ungefähr 0.03 Fälle/1'000 Patientenjahre, mit ungefähr 0.015 Todesfällen/1'000 Patientenjahre). In klinischen Studien mit mehr als 20'000 Patientenjahren mit Metforminexposition gab es keine Berichte über Laktatazidose. Gemeldete Fälle traten hauptsächlich bei Diabetikern mit schwerer Einschränkung der Nierenfunktion oder chronischem Nierenversagen auf. Für Patienten mit Herzinsuffizienz, die eine medikamentöse Behandlung benötigen, im Speziellen für Patienten mit instabiler oder akuter Herzinsuffizienz mit Hypoperfusion und Hypoxie, besteht ein erhöhtes Risiko einer Laktatazidose. Das Risiko einer Laktatazidose steigt mit dem Grad der Niereninsuffizienz und dem Alter des Patienten. Dieses Risiko kann deshalb signifikant gesenkt werden, indem die Nierenfunktion bei Patienten mit Metformintherapie regelmässig kontrolliert wird und die minimale wirksame Metformindosis verwendet wird. Besonders während der Behandlung von älteren Patienten sollte die Nierenfunktion sorgfältig überwacht werden (siehe Verwendung bei älteren Personen, Metforminhydrochlorid). Weiter sollte Metformin bei jeglichen Zuständen, die mit Hypoxie, Dehydrierung und Sepsis assoziiert sind, nicht eingesetzt werden. Da eine gestörte Leberfunktion den Abbau von Laktat signifikant verlangsamen kann, sollte eine Metformintherapie generell bei allen Patienten vermieden werden, bei denen klinisch oder labortechnisch ein Verdacht auf eine Lebererkrankung besteht.
Die Patienten sollten bei der Einnahme von Metformin vor übermässigem akutem oder chronischem Alkoholgenuss gewarnt werden, denn Alkohol verstärkt die Wirkungen von Metforminhydrochlorid auf den Laktatmetabolismus. Zudem sollte Metformin vor intravasaler Gabe von Kontrastmitteln und vor chirurgischen Eingriffen temporär abgesetzt werden.
Der Beginn einer Laktatazidose ist oft subtil und primär von unspezifischen Symptomen, wie beispielsweise Unwohlsein, Myalgien, Atemnot, zunehmender Schläfrigkeit und nichtspezifischen Unterleibsschmerzen begleitet. Bei einer ausgeprägteren Azidose kann es zu Hypothermie, Hypotonie und resistenter Bradyarrhythmie kommen. Der Patient und der behandelnde Arzt müssen sich der möglichen Bedeutung solcher Symptome bewusst sein und der Patient muss instruiert werden, sofort den Arzt zu benachrichtigen, wenn diese Symptome auftreten. Metformin muss in einem solchen Fall abgesetzt und der Patient hospitalisiert werden. Serumelektrolyte und -ketone, Blutzuckerspiegel und der Blut pH-Wert sowie der Laktat- und der Metforminspiegel können bei der Abklärung nützlich sein.
Bei Patienten, die Metformin einnehmen, weisen über den Normalwerten liegende Nüchternwerte von Plasmalaktat, die aber tiefer als 5 mmol/l sind, nicht unbedingt auf eine bevorstehende Laktatazidose hin und können mit anderen Mechanismen erklärt werden, wie beispielsweise mit einem schlecht kontrollierten Diabetes oder Übergewicht, mit intensiver körperlicher Betätigung oder technischen Problemen bei der Handhabung der Proben.
Laktatazidose sollte bei jedem Diabetespatienten mit metabolischer Azidose in Betracht gezogen werden, wenn Hinweise auf eine Ketoazidose fehlen (Ketonurie und Ketonämie).
Laktatazidose ist ein medizinischer Notfall, der in einem Spital behandelt werden muss. Bei einem Patienten mit Laktatazidose, der Metformin einnimmt, sollte das Medikament sofort abgesetzt und die üblichen unterstützenden Massnahmen ergriffen werden. Weil Metforminhydrochlorid und Laktat dialysierbar sind (mit einer Clearance von bis zu 170 ml/min unter guten hämodynamischen Bedingungen), wird eine sofortige Hämodialyse empfohlen. Eine solche Behandlung bewirkt oft eine rasche Behebung der Symptome und eine deutliche Erholung (siehe KONTRAINDIKATIONEN).
Wenn der Patient einmal auf Metformin stabilisiert ist, kommen gastrointestinale Symptome wegen Metformin, die zu Beginn einer Therapie oft auftreten, kaum mehr vor. Wenn später noch gastrointestinale Symptome auftreten, könnte die Ursache eine Laktatazidose oder eine andere ernsthafte Krankheit sein.
Hypoglykämie: Bei Patienten, die Metformin als Monotherapie erhalten, tritt bei normaler Anwendung keine Hypoglykämie auf. In Ausnahmefällen kann im Zusammenhang mit Metformin Hypoglykämie auftreten, z.B. bei unzureichender Kalorienzufuhr oder stark erhöhtem Energieverbrauch (grosser körperlicher Anstrengung) sowie bei gleichzeitiger Einnahme von Alkohol oder anderen glukosesenkenden Substanzen (wie beispielsweise Sulfonylharnstoffen und Insulin).
Ältere, schwache oder schlecht ernährte Patienten und solche mit Nebennieren- oder Hypophyseninsuffizienz oder Alkoholintoxikation, sind besonders anfällig auf hypoglykämische Wirkungen. Hypoglykämie ist bei älteren Personen und bei Menschen, die β-adrenerge Rezeptorenblocker zu sich nehmen, manchmal schwierig zu erkennen.
Gleichzeitige Einnahme von anderen Arzneimitteln, welche die Nierenfunktion oder die Verfügbarkeit von Metformin beeinflussen können: Begleitmedikationen, welche die Nierenfunktion beeinflussen können, eine signifikante Veränderung der Hämodynamik bewirken oder die Verfügbarkeit von Metformin beeinflussen, wie beispielsweise kationische Arzneimittel, die durch renale tubuläre Sekretion ausgeschieden werden (siehe INTERAKTIONEN, Metforminhydrochlorid), sollten mit Vorsicht verwendet werden.
Anwendung von iodhaltigen Kontrastmitteln: Die intravaskuläre Applikation von iodhaltigen Kontrastmitteln für Röntgenuntersuchungen kann zu Nierenversagen führen. Da dies zu Metforminakkumulation und Laktatazidose führen kann, soll die Behandlung mit Metformin bei Patienten mit einer eGFR <60 ml/min/1.73 m2 rechtzeitig vor einer solchen Untersuchung (ca. 2 Tage) unterbrochen werden und erst wiederaufgenommen werden, wenn sich die Nierenfunktion 2 Tage im Anschluss an die Kontrastmitteluntersuchung nicht verschlechtert.
Hypoxie: Herz-Kreislaufversagen verschiedenen Ursprungs, akute Herzinsuffizienz und akuter Herzinfarkt, sowie andere Zustände bei denen Hypoxie auftritt, wurden mit Laktatazidose in Zusammenhang gebracht. Diese Zustände können auch eine prärenale Azotämie verursachen. Wenn solche Vorfälle während der Therapie mit Janumet XR auftreten, muss das Arzneimittel sofort abgesetzt werden.
Operationen: Janumet XR sollte bei chirurgischen Eingriffen temporär abgesetzt werden (ausser bei kleineren Eingriffen, bei denen die Einnahme von Essen und Flüssigkeit unbeschränkt möglich ist) und Janumet XR sollte erst wieder verabreicht werden, wenn eine normale Nahrungsaufnahme wieder möglich ist und die Nierenfunktion als hinreichend eingestuft wurde (siehe DOSIERUNG/ANWENDUNG).
Alkoholkonsum: Es ist bekannt, dass Alkohol die Wirkung von Metformin auf den Laktatmetabolismus verstärkt. Patienten, die Janumet XR einnehmen, müssen deshalb vor exzessivem Alkoholkonsum, akut oder chronisch, gewarnt werden.
Natrium: Dieses Arzneimittel enthält weniger als 1 mmol (23 mg) Natrium pro Tablette und gilt im Wesentlichen als «natriumfrei».
Leberinsuffizienz: Da Leberinsuffizienz in einigen Fällen mit Laktatazidose in Verbindung gebracht wurde, sollte Janumet XR bei Patienten mit klinischem oder labormässigem Verdacht auf eine Lebererkrankung nicht verwendet werden.
Vitamin B12 Blutspiegel: In kontrollierten klinischen Studien mit Metformin während 29 Wochen wurde bei ca. 7% der Patienten eine Verminderung des vorher normalen Vitamin B12 Blutspiegels auf unterdurchschnittliche Werte beobachtet, jedoch ohne klinische Manifestationen. Eine solche Verminderung des B12 Blutspiegels, vermutlich wegen einer Reduktion der B12 Absorption, ist jedoch sehr selten mit einer Anämie assoziiert und scheint schnell reversibel zu sein, sobald Metformin abgesetzt oder zusätzliches Vitamin B12 verabreicht wird. Für alle Patienten, die Janumet XR einnehmen, wird eine jährliche Messung der hämatologischen Parameter empfohlen und alle Abweichungen müssen gründlich untersucht und behandelt werden.
Personen mit inadäquater Zufuhr oder Absorption von Vitamin B12 oder Calcium scheinen prädisponiert, erniedrigte Vitamin B12 Blutspiegel aufzuweisen. Bei diesen Patienten ist alle 2 bis 3 Jahre eine routinemässige Vitamin B12 Bestimmung im Serum angezeigt.
Änderungen im klinischen Status bei Patienten, deren Diabetes Typ 2 vorher unter Kontrolle war: Ein Patient, dessen Diabetes Typ 2 mit Janumet XR vorher gut kontrolliert war, der aber plötzlich anormale Laborwerte aufweist oder klinisch krank wird (oft vage und schlecht definierte Erkrankungen), sollte sofort auf eine mögliche Ketoazidose oder Laktatazidose untersucht werden. Die Untersuchung sollte die Serumelektrolyte und -ketone, den Blutzuckerspiegel und, falls indiziert, den Blut-pH-Wert, den Laktat-, Pyruvat- und Metforminspiegel umfassen. Bei Verdacht auf metabolische Azidose soll – unabhängig von der Genese – Janumet XR sofort abgesetzt und der Patient hospitalisiert werden.
Verlust der Kontrolle über den Blutzuckerspiegel: Bei mit einem beliebigen Antidiabetikum stabil eingestellten Patienten kann in Stresssituationen wie Fieber, Trauma, Infektion oder Operation, temporär eine Entgleisung des Blutzuckerspiegels auftreten. In solchen Fällen kann es vielleicht nötig sein, Janumet XR abzusetzen und temporär durch Insulin zu ersetzen. Janumet XR kann dann wieder verabreicht werden, wenn die akute Episode vorbei ist.
Verwendung bei Kindern und Jugendlichen
Die Sicherheit und Wirksamkeit von Janumet XR und einem seiner Wirkstoffe, Sitagliptin, wurde bei Kindern und Jugendlichen unter 18 Jahren nicht untersucht.
Verwendung bei älteren Personen
Janumet XR
Sitagliptin und Metformin werden hauptsächlich durch die Niere ausgeschieden. Janumet XR sollte daher mit zunehmendem Alter des Patienten mit Vorsicht angewendet werden. Eine Kontrolle der Nierenfunktion ist besonders bei älteren Patienten zur Verhütung der metformin-assoziierten Laktatazidose notwendig (siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN, Metforminhydrochlorid, Laktatazidose).
Sitagliptinphosphat
In klinischen Studien waren Sicherheit und Wirksamkeit von Sitagliptin bei älteren Patienten (≥65 Jahre) ähnlich wie bei jüngeren Personen (<65 Jahre).
Metforminhydrochlorid
An kontrollierten klinischen Studien mit Metformin nahmen nicht genügend ältere Patienten teil, um mit Sicherheit sagen zu können, ob sie anders reagieren als jüngere Patienten. Allerdings zeigen andere klinische Erfahrungen keine Unterschiede zwischen älteren und jüngeren Patienten.
Janumet XR soll grundsätzlich zusammen mit dem Abendessen eingenommen werden. Bei gleichzeitiger Einnahme mit dem Essen können die gastrointestinalen Nebenwirkungen von Metformin teilweise reduziert werden. Ausserdem ist damit zu rechnen, dass die Bioverfügbarkeit von Metformin mit verzögerter Freisetzung bei Einnahme auf nüchternen Magen reduziert ist. Dadurch könnte die Einstellung des Diabetes erschwert werden. Zu beachten ist, dass die Bioverfügbarkeit von Metformin mit nicht verzögerter Freisetzung durch gleichzeitige Nahrungsaufnahme reduziert wird - dies im Gegensatz zu Metformin mit verzögerter Freisetzung.
Interaktionen
Sitagliptin und Metformin
Bei Patienten mit Diabetes Typ 2 beeinflusste die Kombination von multiplen Dosen Sitagliptin (50 mg b.i.d.) und Metformin (1000 mg b.i.d.) weder die Pharmakokinetik von Sitagliptin noch diejenige von Metformin wesentlich.
Pharmakokinetische Interaktionsstudien mit Janumet XR wurden keine durchgeführt. Jedoch wurden solche Studien mit den einzelnen Komponenten von Janumet XR, Sitagliptin und Metformin, durchgeführt.
Sitagliptinphosphat
In pharmakologischen Interaktionsstudien zeigte Sitagliptin keine klinisch signifikante Beeinflussung der Pharmakokinetik folgender Arzneimittel: Metformin, Rosiglitazon, Glibenclamid, Simvastatin, Warfarin und orale Kontrazeptiva. Basierend auf diesen Daten hemmt Sitagliptin die CYP-Isoenzyme CYP3A4, 2C8 und 2C9 nicht. Basierend auf in vitro Daten wird ebenfalls nicht erwartet, dass Sitaglitpin CYP2D6, 1A2, 2C19 oder 2B6 hemmt oder CYP3A4 induziert.
In populations-pharmakokinetischen Analysen mit Typ 2 Diabetikern wurde kein klinisch signifikanter Einfluss der Begleitmedikamentationen auf die Pharmakokinetik von Sitagliptin festgestellt. Untersucht wurden für Typ 2 Diabetiker übliche Arzneimittel.
Bei der gleichzeitigen Verabreichung von Sitaglitpin und Digoxin war ein leichter Anstieg der Fläche unter der Kurve (AUC, 11%) sowie der durchschnittlichen maximalen Plasmakonzentration (Cmax, 18%) von Digoxin zu beobachten. Diese Veränderungen sind klinisch nicht signifikant. Patienten, die Digoxin einnehmen, sollten entsprechend überwacht werden.
Bei gleichzeitiger Verabreichung einer einmaligen, oralen 100 mg Dosis Sitagliptin und einer einmaligen, oralen 600 mg Dosis Cyclosporin, einem potenten Inhibitor des p-Glykoproteins, waren die AUC und Cmax von Sitaglitptin ca. 29% bzw. 68% erhöht. Die beobachteten Veränderungen der Pharmakokinetik von Sitagliptin werden als klinisch nicht signifikant eingestuft.
Metforminhydrochlorid
Glibenclamid: In einer Interaktionsstudie mit einmaliger Verabreichung an Patienten mit Diabetes Typ 2, ergab die gleichzeitige Einnahme von Metformin und Glibenclamid weder eine Veränderung der Pharmakokinetik noch der Pharmakodynamik von Metformin. Bei Glibenclamid wurden erniedrigte AUC- und Cmax-Werte beobachtet, aber diese waren stark variabel. Aufgrund der einmaligen Verabreichung der Arzneimittel in dieser Studie und aufgrund der fehlenden Korrelation zwischen Glibenclamid-Blutspiegel und pharmakodynamischer Wirkung ist keine klare Aussage über die klinische Signifikanz dieser Interaktion möglich.
Furosemid: In einer Interaktionsstudie mit einmaliger Verabreichung von Metformin und Furosemid an gesunde Probanden wurde gezeigt, dass die pharmakokinetischen Parameter beider Arzneimittel beeinflusst wurden. Furosemid erhöhte die Cmax von Metformin im Plasma und Blut um 22% und die Metformin-AUC im Blut um 15%, ohne signifikante Beeinflussung der renalen Clearance von Metformin. Bei der gleichzeitigen Verabreichung von Metformin waren Cmax und AUC von Furosemid im Vergleich zur Monotherapie 31% bzw. 12% erniedrigt und die terminale Halbwertzeit war 32% kürzer, ohne signifikante Beeinflussung der renalen Clearance von Furosemid. Über die Interaktion von Metformin und Furosemid bei längerer Anwendungsdauer sind keine Daten vorhanden.
Nifedipin: In einer Interaktionsstudie mit einmaliger Verabreichung von Metformin und Nifedipin an freiwillige Probanden wurde gezeigt, dass die gleichzeitige Verabreichung von Nifedipin die Cmax und AUC von Metformin im Plasma um 20% bzw. 9% erhöhte, und dieses vermehrt im Urin ausgeschieden wurde. Tmax und die Halbwertzeit wurden nicht beeinflusst. Nifedipin scheint die Absorption von Metformin zu beschleunigen. Metformin beeinflusst Nifedipin kaum.
Die gleichzeitige Anwendung von Arzneimitteln, die mit den tubulären Transportsystemen in der Niere interferieren, welche an der renalen Eliminierung von Metformin beteiligt sind (z.B. organischer Kationentransporter-2 [OCT2]/Multidrug and toxin extrusion [MATE] Inhibitoren, wie Ranolazin, Vandetanib, Dolutegravir und Cimetidin), könnte die systemische Exposition von Metformin und damit das Risiko für eine Laktatazidose erhöhen. In Interaktionsstudien mit einmaliger sowie multipler Verabreichung von Metformin in Kombination mit Cimetidin an gesunden Probanden, erhöhten sich die maximalen Plasma- und Gesamtblutkonzentrationen von Metformin um 60% und die Plasma-AUC und Gesamtblut-AUC von Metformin um 40%. Der Nutzen und die Risiken der gemeinsamen Anwendung dieser Substanzen mit Metformin sind daher abzuwägen.
Andere: Gewisse Arzneimittel können eine Hyperglykämie auslösen und so die Blutzuckereinstellung stören. Solche Arzneimittel sind beispielsweise die Thiazide und andere Diuretika, Kortikosteroide, Phenothiazine, Schilddrüsenarzneimittel, Östrogene, orale Verhütungsmittel, Phenytoin, Nikotinsäure, Sympathomimetika, Calcium-Kanalblocker und Isoniazid. Wenn eines dieser Arzneimittel einem Patienten verabreicht wird, der Janumet XR einnimmt, sollte der Patient engmaschig überwacht werden, um eine adäquate Blutzuckereinstellung zu garantieren.
In Interaktionsstudien mit einmaliger Verabreichung wurde bei gesunden Probanden die Pharmakokinetik von Metformin und Propranolol sowie von Metformin und Ibuprofen nicht beeinflusst, wenn diese jeweils kombiniert verabreicht wurden.
Metformin wird kaum an Plasmaproteine gebunden und interagiert daher kaum mit stark proteingebundenen Arzneimitteln, wie beispielsweise Salicylaten, Sulfonamiden, Chloramphenicol und Probenecid, ganz im Gegensatz zu den Sulfonylharnstoffen, die stark an Serumproteine binden.
Schwangerschaft/Stillzeit
Verwendung während der Schwangerschaft
Es wurden keine adäquaten und streng kontrollierten Studien mit Janumet XR oder dessen einzelnen Inhaltsstoffen bei schwangeren Frauen durchgeführt.
Deshalb ist die Sicherheit von Janumet XR für schwangere Frauen nicht bekannt. Die Einnahme von Janumet XR wird, wie diejenige anderer oraler Antidiabetika, bei Schwangerschaft nicht empfohlen.
Verwendung während der Stillzeit
Es wurden keine Studien mit den kombinierten Wirkstoffen von Janumet XR an laktierenden Tieren durchgeführt. In Studien mit den einzelnen Wirkstoffen wurden sowohl Sitagliptin als auch Metformin in der Milch von laktierenden Ratten ausgeschieden. Es ist nicht bekannt, ob Sitagliptin in der menschlichen Muttermilch ausgeschieden wird. Deshalb sollte Janumet XR von stillenden Frauen nicht eingenommen werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Es wurden keine Studien über die Auswirkungen von Janumet XR auf die Fahrtüchtigkeit und das Bedienen von Maschinen durchgeführt. Es wird jedoch erwartet, dass Janumet XR die Fahrtüchtigkeit und das Bedienen von Maschinen nicht beeinflusst.
Unerwünschte Wirkungen
Es wurden keine klinischen Studien mit Janumet XR Tabletten durchgeführt. Bioäquivalenz von Janumet XR mit gleichzeitig gegebenem Sitagliptin und Metformin mit verzögerter Freisetzung wurde hingegen nachgewiesen (siehe PHARMAKOKINETIK).
In placebokontrollierten klinischen Studien wurde die Kombinationstherapie mit Sitagliptin und Metformin mit nicht verzögerter Freisetzung von Patienten mit Diabetes Typ 2 im Allgemeinen gut vertragen. Die Gesamthäufigkeit von unerwünschten Wirkungen bei Patienten, die eine Kombinationstherapie mit Sitagliptin und Metformin erhielten, war ähnlich wie bei der Einnahme von Placebo und Metformin.
Die Häufigkeiten sind definiert als: häufig ≥1/100, <1/10; gelegentlich ≥1/1000, <1/100; selten ≥1/10000, <1/1000; sehr selten <1/10000.
Kombinationstherapie mit Sitagliptin und Metformin
In einer 24-wöchigen, placebokontrollierten Studie bei Patienten, die mit Diät und Bewegung allein ungenügend eingestellt waren, wurde die Behandlung mit Sitagliptin 50 mg zweimal täglich in Kombination mit Metformin 500 mg oder 1000 mg zweimal täglich untersucht. Die unerwünschten Wirkungen im Zusammenhang mit der Kombinationstherapie, die bei ≥1% der Patienten auftraten (und im Vergleich zur Placebokontrollgruppe häufiger vorkamen) waren wie folgt:
Stoffwechsel- und Ernährungsstörungen:
Häufig: Hypoglykämie.
Störungen des Nervensystems
Häufig: Kopfschmerzen.
Gastrointestinale Beschwerden
Häufig: Diarrhöe, Nausea, Dyspepsie, Flatulenz, Erbrechen.
In einer 24-wöchigen placebokontrollierte Studie mit Sitagliptin, welches zusätzlich zu einer bereits begonnenen Metformin-Monotherapie verabreicht wurde, behandelte man 464 Metformin-Patienten mit 100 mg Sitagliptin einmal täglich und 237 weitere Patienten erhielten Placebo zusammen mit Metformin.
Die unerwünschten Wirkungen im Zusammenhang mit der Kombinationstherapie, die bei ≥1% der Patienten auftraten (und im Vergleich zur Placebokontrollgruppe häufiger vorkamen) waren wie folgt:
Gastrointestinale Beschwerden
Häufig: Nausea.
Hypoglykämie und gastrointestinale Beschwerden
In den placebokontrollierten Studien mit Sitagliptin und Metformin als Kombinationstherapie war die Häufigkeit einer Hypoglykämie (unabhängig von der Kausalitätsbeurteilung durch den Studienarzt) im Zusammenhang mit der Einnahme von Sitagliptin in Kombination mit Metformin etwa gleich gross wie diejenige mit Metformin in Kombination mit Placebo.
Die Häufigkeit gastrointestinaler Beschwerden bei Patienten, die mit der Kombination von Sitagliptin und Metformin behandelt wurden, war vergleichbar mit derjenigen bei Patienten, die mit einer Metformin-Monotherapie behandelt wurden (siehe Tabelle 1).
Tabelle 1:
Hypoglykämie und gastrointestinale Beschwerden (unabhängig von der Kausalitätsbeurteilung durch den Studienarzt), die bei Patienten mit der Kombinationstherapie beobachtet wurden
Anzahl Patienten (%) | ||||||
Studie mit Sitagliptin und Metformin bei unter Diät und Bewegung ungenügend eingestellten Patienten | Studie mit Sitagliptin als Add-on zu Metformin | |||||
Placebo | Sitagliptin 100 mg q.d. | Metformin 500 oder 1000 mg b.i.d. †† | Sitagliptin 50 mg b.i.d. + | Placebo und Metformin ≥1500 mg täglich | Sitagliptin 100 mg q.d. und Metformin ≥1500 mg täglich | |
N= 176 | N= 179 | N= 364 | N= 372 | N= 237 | N= 464 | |
Hypoglykämie | 1 (0.6) | 1 (0.6) | 3 (0.8) | 6 (1.6) | 5 (2.1) | 6 (1.3) |
Diarrhöe | 7 (4.0) | 5 (2.8) | 28 (7.7) | 28 (7.5) | 6 (2.5) | 11 (2.4) |
Nausea | 2 (1.1) | 2 (1.1) | 20 (5.5) | 18 (4.8) | 2 (0.8) | 6 (1.3) |
Erbrechen | 1 (0.6) | 0 (0.0) | 2 (0.5) | 8 (2.1) | 2 (0.8) | 5 (1.1) |
Bauchschmerzen† | 4 (2.3) | 6 (3.4) | 14 (3.8) | 11(3.0) | 9 (3.8) | 10 (2.2) |
† In der Studie bei mit Diät und Bewegung ungenügend eingestellten Patienten wurden Abdominalbeschwerden auch zu den Bauchschmerzen gezählt.
†† Daten gepoolt für Patienten, denen tiefere bzw. höhere Dosen Metformin verabreicht wurden.
In allen Studien wurden alle als «Hypoglykämie» bezeichneten Nebenwirkungen berücksichtigt, d.h. auch solche, die nicht durch eine Blutzuckerbestimmung bestätigt wurden.
Sitagliptin in Kombination mit Metformin und einem Sulfonylharnstoff
In einer 24-wöchigen, placebokontrollierten Studie mit 100 mg Sitagliptin täglich, welches zusätzlich zu einer bereits begonnenen Glimepirid-Dosis von ≥4 mg täglich und Metformin ≥1500 mg täglich verabreicht wurde, waren die arzneimittelbedingten unerwünschten Wirkungen, die bei ≥1% der Sitagliptin-Patienten beobachtet wurden (N=116) und häufiger auftraten als bei der Placebokontrollgruppe (N=113) die folgenden:
Stoffwechsel- und Ernährungsstörungen
Häufig: Hypoglykämie.
Gastrointestinale Beschwerden
Häufig: Obstipation.
Während der Behandlung mit Sitagliptin in Kombination mit Metformin wurden keine klinisch signifikanten Änderungen bei den Vitalfunktionen oder beim EKG (einschliesslich QT-Intervall) beobachtet.
Sitagliptin in Kombination mit Metformin und Insulin
In einer 24-wöchigen, placebokontrollierten Studie mit Sitagliptin 100 mg zusätzlich zu einer bestehenden Kombinationsbehandlung mit Metformin ≥1500 mg täglich und einer stabilen Dosis Insulin, war Hypoglykämie die einzige unerwünschte Reaktion, welche bei ≥1% der Patienten unter Sitagliptin (N=229) festgestellt wurde und die häufiger war als bei Patienten unter Placebo (N=233) (Sitagliptin 15.3%; Placebo 8.2%). Bei diesen Häufigkeitsangaben wurden alle Nebenwirkungen berücksichtigt, unabhängig von der Kausalitätsbeurteilung durch den Studienarzt. Insbesondere bei den Hypoglykämieraten ist zu berücksichtigen, dass alle Patienten neben Sitagliptin oder Placebo auch mit Insulin behandelt wurden, teils zusätzlich mit Metformin. In einer anderen 24-wöchigen Studie mit Patienten, die während einer Intensivierung des Insulins (mit oder ohne Metformin) Sitagliptin als Add-on-Therapie erhielten, waren die unerwünschten Wirkungen, die bei ≥1% der mit Sitagliptin und Metformin behandelten Patienten (N=285) festgestellt wurden und häufiger waren als bei mit Placebo und Metformin behandelten Patienten (N=283): Obstipation (Sitagliptin und Metformin, 1.4%; Placebo und Metformin, 1.1%), Diarrhö (4.9%; 2.5%), Erbrechen (3.2%; 1.1%), peripheres Ödem (2.1%; 1.4%), Fieber (1.1%; 0.4%), Bronchitis (1.4%; 1.1%), Zellulitis (1.4%; 1.1%), Pharyngitis (1.8%; 1.1%), Infektion der oberen Atemwege (4.2%; 1.4%), verminderte Kreatininclearance (1.1%; 0.0%), Schmerzen des Muskel- und Skelettsystems (1.4%; 1.1%), Myalgie (1.1%; 0.7%), Nephrolithiasis (1.1%; 0.4%) und Husten (2.8%; 1.8%). Bei diesen Häufigkeitsangaben wurden alle unerwünschten Wirkungen berücksichtigt, unabhängig von der Kausalitätsbeurteilung durch die Prüfärzte. Zudem lag die Hypoglykämie-Inzidenz bei 24.9% für mit Sitagliptin, Metformin und Insulin behandelte Patienten und bei 37.8% für Patienten unter Placebo, Metformin und Insulin.
Bekannte unerwünschte Wirkungen mit Sitagliptin
Bei Patienten, die Sitagliptin einnahmen, wurden keine arzneimittelbedingten, unerwünschten Wirkungen mit einer Häufigkeit von ≥1% beobachtet.
Bekannte unerwünschte Wirkungen mit Metformin
Störungen des Blut- und Lymphsystems
Vereinzelte Fälle von Leukopenie, Thrombopenie und hämolytischer Anämie.
Sehr selten: erniedrigter Vitamin B12 Blutspiegel.
Stoffwechsel- und Ernährungsstörungen
Sehr selten: Laktatazidose (Inzidenz 3 Fälle/100'000 Patientenjahre, vgl. WARNHINWEISE UND VORSICHTSMASSNAHMEN).
Störungen des Nervensystems
Häufig: Metallgeschmack (3%).
Gastrointestinale Beschwerden
Häufig: Gastrointestinale Störungen wie z.B. Nausea, Erbrechen, Diarrhöe, Bauchschmerzen, Appetitverlust.
Diese Symptome treten meist zu Beginn der Therapie auf und gehen in der Regel spontan zurück.
Funktionsstörungen der Leber und Galle
In Einzelfällen: abnorme Werte in Leberfunktionstests, z.B. erhöhte Transaminasen oder Hepatitis (nach Absetzen von Metformin reversibel).
Funktionsstörungen der Haut und des Unterhautzellgewebes
Sehr selten: Hautreaktionen wie Erythem, Pruritus, Urtikaria.
TECOS kardiovaskuläre Sicherheitsstudie: Die TECOS-Studie (Trial Evaluating Cardiovascular Outcomes with Sitagliptin) umfasste eine Intention-to-treat Population, von denen 7332 Patienten täglich mit 100 mg Sitagliptin (bzw. 50 mg täglich, falls die geschätzte glomeruläre Filtrationsrate (eGFR) zu Studienbeginn zwischen ≥30 und <50 ml/min/1.73m2 lag) behandelt wurden und 7339 Patienten mit Placebo behandelt wurden. Beide Behandlungen wurden zur üblichen Therapie hinzugefügt. Die Studienpopulation beinhaltete total 2004 Patienten, die ≥75 Jahre alt waren (970 mit Sitagliptin und 1034 mit Placebo behandelt). Die Häufigkeit von schwerwiegenden unerwünschten Ereignissen war insgesamt bei Patienten unter Sitagliptin vergleichbar mit den Patienten unter Placebo. Eine Beurteilung der vordefinierten Diabetes-bezogenen Komplikationen ergab ähnliche Inzidenzen zwischen den Gruppen, einschliesslich Infektionen (18.4% der mit Sitagliptin behandelten Patienten und 17.7% der mit Placebo behandelten Patienten) und Nierenversagen (1.4% der mit Sitagliptin behandelten Patienten und 1.5% der mit Placebo behandelten Patienten). Das Profil der unerwünschten Ereignisse bei Patienten ≥75 Jahre war im Allgemeinen ähnlich zur Gesamtpopulation.
Bei Patienten, welche zum Studienbeginn Insulin und/oder einen Sulfonylharnstoff verwendeten, betrug die Inzidenz einer schweren Hypoglykämie 2.7% bei mit Sitagliptin behandelten Patienten und 2.5% bei mit Placebo behandelten Patienten. Bei Patienten welche zu Studienbeginn kein Insulin und/oder einen Sulfonylharnstoff verwendeten, betrug die Inzidenz einer schweren Hypoglykämie 1.0% bei mit Sitagliptin behandelten Patienten und 0.7% bei mit Placebo behandelten Patienten. Die Inzidenz von Pankreatitiden, die durch ein Nachprüfungskommitee bestätigt wurden, betrug 0.3% bei mit Sitagliptin behandelten Patienten und 0.2% bei mit Placebo behandelten Patienten. Die Inzidenz von Malignomen, die durch ein Nachprüfungskommitee bestätigt wurden, betrug 3.7% bei mit Sitagliptin behandelten Patienten und 4.0% bei mit Placebo behandelten Patienten.
Pankreatitis: In einer gepoolten Analyse von 19 doppelblinden klinischen Studien an 10'246 Patienten randomisiert für 100 mg Sitagliptin täglich (N = 5429) oder entsprechende Kontrolle (aktiv oder Placebo) (N = 4817) war die Inzidenz akuter Pankreatitiden in jeder Gruppe 0.1 pro 100 Patientenjahre (für Sitagliptin 4 Patienten mit einem Ereignis in 4708 Patientenjahren; für die Kontrolle 4 Patienten mit einem Ereignis in 3942 Patientenjahren) (siehe auch TECOS kardiovaskuläre Sicherheitsstudie und WARNHINWEISE UND VORSICHTSMASSNAHMEN, Pankreatitis).
Postmarketing Erfahrungen
Zusätzliche unerwünschte Reaktionen wurden nach Markteinführung von Janumet XR oder Sitagliptin, einer der Komponenten von Janumet XR, gemeldet. Beim alleinigen Gebrauch von Janumet XR oder Sitagliptin und/oder zusammen mit anderen antihyperglykämischen Substanzen wurde über diese unerwünschten Wirkungen berichtet. Da diese auf Spontanmeldungen einer unbekannt grossen Population beruhen, ist es nicht möglich die Häufigkeit oder Kausalität zu bestimmen: Überempfindlichkeitsreaktionen einschliesslich Anaphylaxie, Angioödem, Hautausschlag, Urticaria, kutane Vaskulitis und exfoliative Hautveränderungen, einschliesslich Stevens-Johnson Syndrom (siehe KONTRAINDIKATIONEN und WARNHINWEISE UND VORSICHTSMASSNAHMEN, Überempfindlichkeitsreaktionen); akute Pankreatitis einschliesslich hämorrhagische Pankreatitis mit fatalem und nicht-fatalem Ausgang und nekrotisierende Pankreatitis (siehe auch WARNHINWEISE UND VORSICHTSMASSNAHMEN); Verschlechterung der Nierenfunktion einschliesslich akutes Nierenversagen (in gewissen Fällen ist Dialyse erforderlich); Rhabdomyolyse; bullöses Pemphigoid (siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN, Bullöses Pemphigoid); Infektionen des oberen Respirationstrakts, Nasopharyngitis; Obstipation, Erbrechen; Kopfschmerzen; Arthralgie, Myalgie, Schmerzen in den Extremitäten, Rückenschmerzen; Pruritus.
Laborwerte
Sitagliptinphosphat
Die Häufigkeit von Veränderungen der Laborparameter war bei den mit Sitagliptin und Metformin behandelten Patienten ähnlich wie bei der Kontrollgruppe, die mit Placebo und Metformin behandelt wurde.
In mehreren klinischen Studien wurde eine leichte Erhöhung der Leukozytenwerte beobachtet (ca. 200 Zellen/µl im Vergleich zu Placebo; mittlerer Ausgangswert der Leukozytenwerte ca. 6600 Zellen/µl) und zwar aufgrund einer leichten Erhöhung der Neutrophilen. Diese Beobachtung wurde in den meisten, aber nicht in allen Studien gemacht. Diese Veränderungen der Laborparameter werden als klinisch nicht relevant betrachtet.
Metforminhydrochlorid
In kontrollierten klinischen Studien mit Metformin während 29 Wochen, wurde bei ca. 7% der Patienten eine Erniedrigung des vorher normalen Vitamin B12 Blutspiegels auf unterdurchschnittliche Werte beobachtet, jedoch ohne klinische Manifestationen.
Eine solche Verminderung des B12 Blutspiegels, vermutlich wegen einer Interferenz mit der B12 Absorption durch den B12-Intrinsic-Faktor-Komplex, ist jedoch sehr selten mit einer Anämie assoziiert und scheint schnell reversibel zu sein, sobald Metformin abgesetzt oder zusätzliches Vitamin B12 verabreicht wird (siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN, Metforminhydrochlorid).
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Sitagliptinphosphat
In kontrollierten klinischen Studien mit gesunden Probanden wurden Einzeldosen von bis zu 800 mg Sitagliptin allgemein gut vertragen.
Bei einer Dosis von 800 mg Sitagliptin wurden in einer Studie minimale Erhöhungen des QT-Intervalls beobachtet, die jedoch als klinisch nicht relevant erachtet wurden.
Es existieren keine Daten für Dosen über 800 mg in klinischen Studien.
In Phase I-Studien mit multipler Verabreichung von Sitagliptin bis zu 600 mg täglich während bis zu 10 Tagen und 400 mg täglich während bis zu 28 Tagen, wurden klinisch keine dosisabhängigen, unerwünschten Wirkungen beobachtet.
Im Fall einer Überdosierung ist es angemessen, die üblichen unterstützenden Massnahmen zu ergreifen, wie z.B. die Entfernung von noch nicht absorbierten Wirkstoffen aus dem Magen-Darm-Trakt, die klinische Überwachung (einschliesslich EKG) und falls nötig die Einleitung einer unterstützenden Therapie.
Sitagliptin ist mässig dialysierbar. In klinischen Studien wurden ca. 13.5% der Dosis während einer 3- bis 4-stündigen Hämodialyse entfernt. Eine längere Hämodialyse kann in Erwägung gezogen werden, falls es klinisch sinnvoll ist. Es ist nicht bekannt, ob Sitagliptin mittels Peritonealdialyse dialysierbar ist.
Metforminhydrochlorid
Überdosierungen von Metformin sind vorgekommen, sogar Überdosierungen über 50 Gramm. Hypoglykämie wurde in ca. 10% der Fälle beobachtet, es konnte jedoch kein kausaler Zusammenhang mit Metformin bewiesen werden. In ca. 32% der Fälle von Überdosierung mit Metformin wurde Laktatazidose festgestellt (siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN, Metforminhydrochlorid). Weil Metformin dialysierbar ist (mit einer Clearance von bis zu 170 ml/min unter guten hämodynamischen Bedingungen), wird eine Hämodialyse empfohlen, um das akkumulierte Metformin zu entfernen.
Eigenschaften/Wirkungen
ATC-Code
A10BD07
Wirkungsmechanismus
Sitagliptinphosphat gehört zu einer Klasse oraler Antidiabetika, den Dipeptidylpeptidase-4 (DPP-4) Inhibitoren, welche die Blutzuckereinstellung bei Patienten mit Diabetes Typ 2 verbessern, indem sie die Konzentrationen der aktiven Inkretine erhöhen. Inkretine, einschliesslich GLP-1 (Glucagon-like Peptide-1) und GIP (Glucose-dependent insulinotropic Polypeptide), werden während des ganzen Tages aus dem Darm freigesetzt und deren Konzentrationen werden durch die Einnahme von Nahrung erhöht. Die Inkretine sind Teil eines endogenen Systems, das bei der physiologischen Regulierung der Glukosehomöostase beteiligt ist. Wenn der Blutzucker normal oder erhöht ist, fördern GLP-1 und GIP die Insulinsynthese und die Insulinfreisetzung aus den Betazellen im Pankreas durch intrazelluläre Signale mit Hilfe von cyclischem AMP. Die Behandlung von Diabetes Typ 2 mit GLP-1 oder mit DPP-4-Inhibitoren in Tiermodellen hat gezeigt, dass die Empfindlichkeit der Betazellen auf Glukose verbessert und die Biosynthese und Freisetzung von Insulin stimuliert wird. Bei höheren Insulinkonzentrationen ist die Glukoseaufnahme des Gewebes erhöht. Zusätzlich erniedrigt GLP-1 die Glukagonsekretion aus den Alphazellen im Pankreas. Erniedrigte Glukagonkonzentrationen, zusammen mit höheren Insulinkonzentrationen, führen zu einer verminderten Glukoseproduktion in der Leber, was wiederum zu einer Erniedrigung des Blutzuckers führt. Die Wirkung von GLP-1 und GIP sind glucoseabhängig. Wenn der Blutzucker tief ist, stimuliert GLP-1 weder die Insulinfreisetzung noch die Suppression der Glukagonsekretion. Durch GLP-1 und GIP wird die Insulinsekretion erhöht wenn Glucose die Normalwerte überschreitet. GLP-1 beeinträchtigt die normale Glukagonreaktion bei einer Hypoglykämie nicht. Die Aktivität von GLP-1 und GIP wird durch das DPP-4-Enzym geregelt, das die Inkretine schnell hydrolysiert und in inaktive Substanzen umwandelt. Sitagliptin verhindert die Hydrolyse der Inkretine durch DPP-4, wobei die Plasmakonzentrationen der aktiven Formen von GLP-1 und GIP erhöht werden. Durch die Konzentrationserhöhung der aktiven Inkretine steigert Sitagliptin die Insulinfreisetzung und erniedrigt die Glukagonkonzentrationen in einer glukoseabhängigen Weise. Bei Diabetes Typ 2-Patienten mit Hyperglykämie führen diese Veränderungen der Insulin- und Glukagonkonzentrationen zu einer geringeren Konzentration des Hämoglobins A1c (HbA1c) sowie tieferen postprandialen und Nüchtern-Blutzuckerspiegeln. Dieser glucoseabhängige Mechanismus unterscheidet sich von demjenigen der Sulfonylharnstoffe, wobei Insulin auch bei niedrigen Glucosewerten freigesetzt wird, was bei Typ 2 Diabetikern und Gesunden zu Hypoglykämien führen kann. Sitagliptin ist ein potenter und stark selektiver Inhibitor des Enzyms DPP-4 und hemmt die nahe verwandten Enzyme DPP-8 und DPP-9 in therapeutischen Konzentrationen nicht.
Metforminhydrochlorid
Metformin ist ein Antidiabetikum, das die Glukosetoleranz bei Patienten mit Diabetes Typ 2 verbessert und sowohl die basalen als auch die postprandialen Plasmaglukosespiegel senkt. Die pharmakologischen Wirkungsmechanismen von Metformin unterscheiden sich von anderen Klassen oraler Antidiabetika. Metformin vermindert sowohl die Glukosesynthese in der Leber als auch die Glukoseabsorption im Darm und verbessert die Insulinsensitivität, indem die periphere Glukoseaufnahme und -verwertung erhöht wird. Im Unterschied zu den Sulfonylharnstoffen erzeugt Metformin weder bei Patienten mit Diabetes Typ 2 noch bei gesunden Probanden eine Hypoglykämie, ausser in sehr speziellen Fällen (siehe WARNHINWEISE UND VORSICHTSMASSNAHMEN, Metforminhydrochlorid) und verursacht auch keine Hyperinsulinämie. Während einer Metformintherapie bleibt die Insulinsekretion unverändert, während der Nüchtern-Insulinspiegel und die Plasmainsulin-Response im Laufe des Tages sogar abnehmen können.
Pharmakodynamik
Nicht zutreffend.
Klinische Wirksamkeit
Klinische Studien
Es wurden keine klinischen Studien zur Wirksamkeit und Sicherheit von Janumet XR durchgeführt. Klinische Studien mit gleichzeitiger Verabreichung von Sitagliptin und Metformin mit nicht verzögerter Freisetzung zeigten signifikante Verbesserungen bei der Blutzuckereinstellung bei Patienten mit Diabetes Typ 2. Die Bioäquivalenz zwischen Janumet XR und gleichzeitig verabreichtem Sitagliptin und Metformin Tabletten mit verzögerter Wirkstofffreisetzung wurde für alle Dosisstärken gezeigt.
In einer Vergleichsstudie war die Verabreichung von Metformin mit verzögerter Wirkstofffreisetzung einmal täglich bei allen Messparametern zur Blutzuckereinstellung ähnlich wirksam, wie die üblicherweise verschriebene zweimal tägliche Verabreichung von Metformin mit nicht verzögerter Wirkstofffreisetzung.
Sitagliptin und Metformin mit nicht verzögerter Freisetzung bei Patienten mit Diabetes Typ 2
An einer randomisierten, doppelblinden, placebokontrollierten Studie zur Untersuchung der Sicherheit und Wirksamkeit der Kombination von Sitagliptin und Metformin nahmen während 24 Wochen insgesamt 1091 Patienten mit Diabetes Typ 2 teil, die trotz Diät und körperlicher Betätigung eine inadäquate Blutzuckereinstellung aufwiesen.
Bei Patienten mit ungenügender glykämischer Kontrolle unter Diät und Bewegung alleine erzielte die Kombination von Sitagliptin und Metformin signifikante Verbesserungen bei HbA1c, FPG (Nüchtern-Plasmaglukose) und 2-Stunden-Wert der PPG im Vergleich zu Placebo, zur Metformin-Monotherapie und zur Sitagliptin-Monotherapie (p<0.001; Tabelle 2, Abbildung 1). Eine Verbesserung mit fast maximaler FPG-Reduktion wurde am ersten Endpunkt nach 3 Wochen erreicht (entsprechend dem ersten Endpunkt nach Beginn der Therapie) und konnte während der ganzen 24-wöchigen Studie aufrechterhalten werden. Im Vergleich zu Placebo zeigten Patienten mit einem höheren HbA1c-Ausgangswert eine stärkere mittlere HbA1c-Reduktion.
Die Verbesserung der HbA1c-Werte wurde durch Geschlecht, Alter, Rasse oder BMI-Ausgangswert nicht beeinflusst. Messungen der Betazellenfunktion, HOMA-β, und das Verhältnis Proinsulin/Insulin zeigten ebenfalls eine Verbesserung, wenn Sitagliptin und Metformin als Kombinationstherapie verabreicht wurden, im Vergleich zu den jeweiligen Monotherapien. Die Lipid-Endpunkte wurden nicht beeinflusst. Die mittleren HbA1c-Reduktionen für die Subgruppe von Patienten ohne Antidiabetika bei Studieneintritt waren: Sitagliptin 100 mg einmal täglich (n=88), -1.06%; Metformin 500 mg zweimal täglich (n=90), -1.09%; Metformin 1000 mg zweimal täglich (n=87), -1.24%; Sitagliptin 50 mg zweimal täglich mit Metformin 500 mg zweimal täglich (n=100), -1.59%; und Sitagliptin 50 mg zweimal täglich mit Metformin 1000 mg zweimal täglich (n=86), -1.94%; und bei Patienten, welche Placebo erhalten haben (n=83), -0.17%.
Abb. 1: Mittlere Veränderung der HbA1c über 24 Wochen gegenüber den Ausgangswerten mit Sitagliptin und Metformin alleine und in Kombination bei Patienten mit Typ-2 Diabetes †
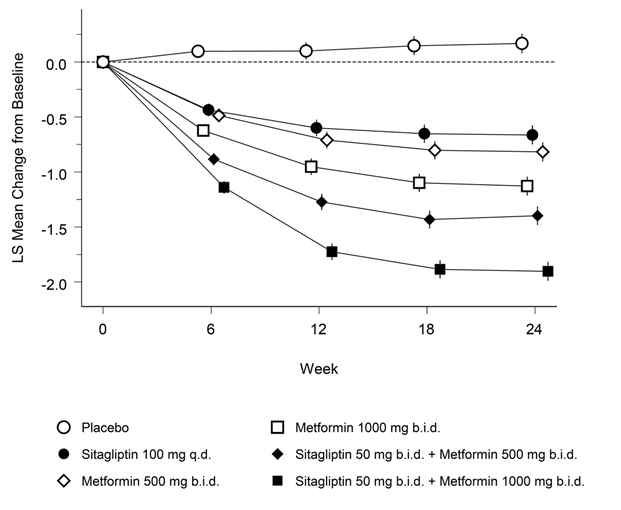
† All Patients Treated Population (eine Intention-to-treat Analyse), Mittelwerte der kleinsten Quadrate, adjustiert für den vorherigen Antidiabetika-Therapiestatus und den Ausgangswert.
Tabelle 2:
Glykämische Parameter und Körpergewicht bei der abschliessenden Untersuchung nach 24 Wochen †
Placebo | Sitagliptin 100 mg q.d. | Metformin 500 mg b.i.d. | Sitagliptin 50 mg b.i.d. | Metformin 1000 mg b.i.d. | Sitagliptin 50 mg b.i.d | |
HbA1c (%) | N = 165 | N = 175 | N = 178 | N = 183 | N = 177 | N = 178 |
Ausgangswert (Mittel) | 8.68 | 8.87 | 8.90 | 8.79 | 8.68 | 8.76 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel‡) | 0.17 | -0.66 | -0.82 | -1.40 | -1.13 | -1.90 |
Veränderung gegenüber Placebo (adjustiertes Mittel‡) | - | -0.83§ | -0.99§ | -1.57§ | -1.30§ | -2.07§ |
Patienten (%) mit HbA1c <7% | 15 (9.1) | 35 (20.0) | 41 (23.0) | 79 (43.2) | 68 (38.4) | 118 (66.3) |
FPG (mmol/l) | N = 169 | N = 178 | N = 179 | N = 183 | N = 179 | N = 180 |
Ausgangswert (Mittel) | 10.91 | 11.19 | 11.40 | 11.33 | 10.94 | 10.93 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel‡) | 0.32 | -0.97 | -1.52 | -2.62 | -1.63 | -3.55 |
Veränderung gegenüber Placebo (adjustiertes Mittel‡) | - | -1.29§ | -1.84§ | -2.94§ | -1.95§ | -3.87§ |
2-Stunden-Wert der PPG (mmol/l) | N = 129 | N = 136 | N = 141 | N = 147 | N = 138 | N = 152 |
Ausgangswert (Mittel) | 15.38 | 15.86 | 16.26 | 16.21 | 15.74 | 15.94 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel‡) | 0.02 | -2.88 | -2.97 | -5.14 | -4.33 | -6.48 |
Veränderung gegenüber Placebo (adjustiertes Mittel‡) | -2.9§ | -2.98§ | -5.16§ | -4.35§ | -6.49§ | |
Körpergewicht (kg)** | N = 167 | N = 175 | N = 179 | N = 184 | N = 175 | N = 178 |
Ausgangswert (Mittel) | 90.1 | 85.9 | 88.1 | 90.0 | 89.4 | 88.2 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel‡) | -0.9 | 0.0 | -0.9 | -0.6 | -1.1 | -1.3 |
Veränderung gegenüber Placebo (adjustiertes Mittel‡) | 0.9¶ | 0.1# | 0.4# | -0.1# | -0.3# |
† All Patients Treated Population (eine Intention-to-treat Analyse).
‡ Mittelwerte der kleinsten Quadrate, adjustiert für den vorherigen Antidiabetika-Therapiestatus und den Ausgangswert.
§ p<0.001 im Vergleich zu Placebo.
** All Patients as Treated (APaT) Population, ohne Daten nach einer glykämischen Notfallbehandlung.
¶ p=0.005 im Vergleich zu Placebo.
# Statistisch nicht signifikant (p≥0.05) im Vergleich zu Placebo.
Zudem schloss diese Studie Patienten (N=117) mit einer starken Hyperglykämie ein (HbA1c >11% oder Blutglukose >15.56 mmol/l), die zweimal täglich mit Sitagliptin 50 mg und Metformin 1000 mg behandelt wurden. Bei diesen Patienten gab es keine Placebokontrolle. In dieser Patientengruppe war der HbA1c-Ausgangswert im Mittel 11.15%, FPG war 17.47 mmol/l und der 2-Stunden-Wert der PPG betrug 24.5 mmol/l. Nach 24 Wochen wurden folgende Reduktionen gegenüber dem Ausgangswert festgestellt: ‑2.9% für HbA1c, ‑7.04 mmol/l für FPG und ‑11.55 mmol/l für den 2-Stunden-Wert der PPG. Nach 24 Wochen wurde in dieser Kohorte ein Anstieg des Körpergewichts um 1.3 kg festgestellt.
Sitagliptin Add-on-Therapie bei Patienten, die mit einer Metformin-Monotherapie eine inadäquate Blutzuckereinstellung haben
In zwei doppelblinden, placebokontrollierten klinischen Studien bei Patienten mit Diabetes mellitus Typ 2 wurden Sitagliptin und Metformin mit nicht verzögerter Freisetzung in Kombination auf Sicherheit und Wirksamkeit geprüft. In beiden Studien wurden Patienten mit inadäquater Blutzuckereinstellung bei gleich bleibenden Dosen Metformin ≥1500 mg randomisiert und erhielten einmal täglich entweder 100 mg Sitagliptin oder Placebo zusätzlich zur bestehenden Therapie mit Metformin.
In einer Studie erhielten 701 Patienten während 24 Wochen einmal täglich 100 mg Sitagliptin oder Placebo. Die Addition von Sitagliptin zur bestehenden Therapie mit Metformin mit nicht verzögerter Freisetzung ergab signifikante Verbesserungen bei HbA1c (-0.65%), FPG (-1.41 mmol/l) und beim 2h-ppBZ (-2.81 mmol/l), im Vergleich zur Addition von Placebo zur bestehenden Therapie mit Metformin (siehe Abb. 2 und Tabelle 3). Die Verbesserung des HbA1C im Vergleich zu Placebo, wurde durch den HbA1C-Ausgangswert, vorhergehende Antidiabetika-Therapien, Geschlecht, Alter, BMI-Ausgangswert, Zeit seit der Diabetesdiagnose, Vorhandensein des metabolischen Syndroms, Standardindizes der Insulinresistenz (HOMA-IR) oder Insulinsekretion (HOMA-β) nicht beeinflusst.
Abb. 2: Mittlere Veränderung des HbA1c-Ausgangswertes in 24 Wochen bei einer täglichen 100 mg-Dosis Sitagliptin und Metformin oder Placebo und Metformin bei Patienten mit Diabetes Typ 2† ‡
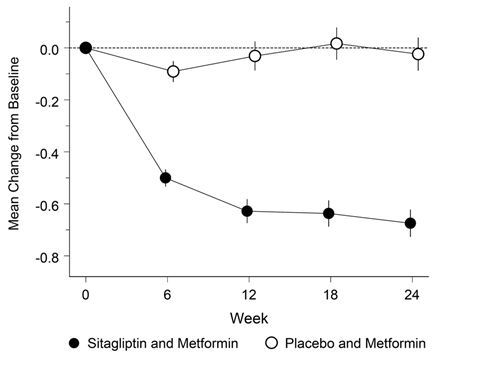
† Patienten mit Metformin-Monotherapie und inadäquater Blutzuckereinstellung.
‡ All Patients Treated Population. Mittelwerte der kleinsten Quadrate, adjustiert für den vorherigen Antidiabetika-Therapiestatus und den Ausgangswert.
Tabelle 3:
Glykämische Parameter und Körpergewicht bei der abschliessenden Untersuchung (24-wöchige Studie)
Sitagliptin als Add-on-Therapie bei Patienten mit inadäquater Blutzuckereinstellung unter Metformin†
Sitagliptin 100 mg q.d. | Placebo | |
HbA1c (%) | N = 453 | N = 224 |
Ausgangswert (Mittel) | 7.96 | 8.03 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel‡) | -0.67 | -0.02 |
Veränderung gegenüber Placebo + Metformin (adjustiertes Mittel‡) | -0.65§ | |
Patienten (%) mit HbA1c <7% | 213 (47.0) | 41 (18.3) |
FPG (mmol/l) | N = 454 | N = 226 |
Ausgangswert (Mittel) | 9.44 | 9.64 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel‡) | -0.94 | 0.47 |
Veränderung gegenüber Placebo + Metformin (adjustiertes Mittel‡) | -1.41§ | |
2-Stunden-Wert der PPG (mmol/l) | N = 387 | N = 182 |
Ausgangswert (Mittel) | 15.25 | 15.13 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel‡) | -3.44 | -0.63 |
Veränderung gegenüber Placebo + Metformin (adjustiertes Mittel‡) | -2.81§ | |
Body Weight (kg)** | N = 399 | N = 169 |
Ausgangswert (Mittel) | 86.9 | 87.6 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel‡) | -0.7 | -0.6 |
Veränderung gegenüber Placebo + Metformin (adjustiertes Mittel‡) | -0.1¶ |
† All Patients Treated Population (eine Intention-to-treat Analyse).
‡ Mittelwerte der kleinsten Quadrate, adjustiert für den vorherigen Antidiabetika-Therapiestatus und den Ausgangswert.
§ p<0.001 im Vergleich zu Placebo + Metformin.
** All Patients as Treated (APaT) Population, ohne Daten nach einer glykämischen Notfallbehandlung.
¶ Statistisch nicht relevant (p≥0.05) im Vergleich zu Placebo + Metformin.
Abb. 3: 24-Stunden Plasmaglukoseprofile bei Patienten mit Diabetes Typ 2 nach 4-wöchiger Behandlung mit Sitagliptin 50 mg BID und Metformin oder Placebo und Metformin†
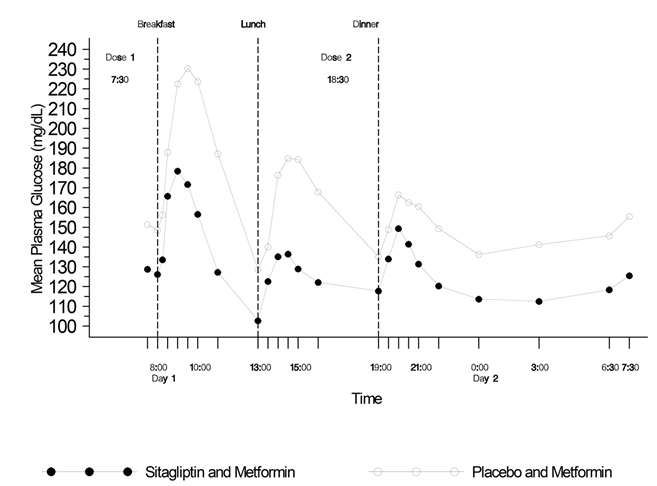
† Patienten mit inadäquater Blutzuckereinstellung mit einer Metformin-Monotherapie.
Sitagliptin Add-on-Therapie bei Patienten, die mit einer Kombination von Metformin mit nicht verzögerter Freisetzung und Glimepirid eine inadäquate Blutzuckereinstellung aufwiesen
Insgesamt 441 Patienten mit Diabetes Typ 2 nahmen während 24 Wochen an einer randomisierten, doppelblinden, placebokontrollierten Studie teil, um die Wirksamkeit von 100 mg Sitagliptin einmal täglich, im Vergleich zu Placebo, in Kombination mit Glimepirid (als Monotherapie oder in Kombination mit Metformin) zu beurteilen. In dieser Studie erhielten 220 Patienten bereits eine Kombinationstherapie mit Glimepirid (≥4 mg pro Tag) und Metformin (≥1500 mg pro Tag).
Die Kombination von Sitagliptin, Glimepirid und Metformin ergab eine signifikante Reduktion des HbA1c-Ausgangswertes (-0.89%) und der FPG (-1.15 mmol/l) im Vergleich zu Placebo (siehe Tabelle 4). Im Vergleich zu Placebo zeigten Patienten mit einem höheren HbA1c-Ausgangswert eine stärkere mittlere HbA1c-Reduktion.
Tabelle 4
Glykämische* Parameter und Körpergewicht bei der abschliessenden Untersuchung (24-wöchige Studie) mit Sitagliptin zusätzlich zur Kombinationstherapie mit Glimepirid und Metformin†
Sitagliptin 100 mg | Placebo | |
HbA1c (%) | N = 115 | N = 105 |
Ausgangswert (Mittel) | 8.27 | 8.28 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel)‡ | -0.59 | 0.30 |
Veränderung gegenüber Placebo (adjustiertes Mittel‡) | -0.89§ | |
Patienten (%) mit HbA1c <7% | 26 (22.6) | 1 (1.0) |
FPG (mmol/l) | N = 115 | N = 109 |
Ausgangswert (Mittel) | 9.96 | 9.94 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel)‡ | -0.43 | 0.72 |
Veränderung gegenüber Placebo (adjustiertes Mittel‡) | -1.15§ | |
Körpergewicht (kg)** | N = 102 | N = 74 |
Ausgangswert (Mittel) | 86.5 | 84.6 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel)‡ | 0.4 | -0.7 |
Veränderung gegenüber Placebo (adjustiertes Mittel‡) | 1.1†† |
* Primäre und sekundäre glykämische Parameter waren HbA1c und Nüchternglucose
† All Patients Treated Population (eine Intention-to-treat Analyse).
‡ Mittelwerte der kleinsten Quadrate, adjustiert für den vorherigen Antidiabetika-Therapiestatus und den Ausgangswert.
§ p<0.001 im Vergleich zu Placebo.
** All Patients as Treated (APaT) Population, ohne Daten nach einer glykämischen Notfallbehandlung.
†† p=0.007 im Vergleich zu Placebo
Sitagliptin Add-on-Therapie bei Patienten, welche unter einer Kombination von Metformin mit nicht verzögerter Freisetzung und Insulin ungenügend kontrolliert sind:
Insgesamt 641 Patienten mit Diabetes Typ 2 nahmen an einer 24-wöchigen, randomisierten, doppelblinden, placebokontrollierten Studie teil, in deren Rahmen die Wirksamkeit von Sitagliptin 100 mg einmal täglich in Kombination mit einer stabilen Dosis Insulin beurteilt wurde. Ungefähr 75% der Patienten nahmen gleichzeitig Metformin ein. Patienten mit fertig hergestelltem lang oder intermediär wirksamem Insulin (mit oder ohne Metformin) wurden zusätzlich entweder zu 100 mg Sitagliptin oder Placebo randomisiert. Gemessene glykämische Endpunkte schlossen HbA1c, Nüchtern-Glucose und 2-Stunden-postprandial-Glucose (PPG) ein.
Die Kombination von Sitagliptin, Metformin und Insulin bewirkte signifikante Verbesserungen des HbA1c, der Nüchtern-Glucose und der 2-Stunden-PPG im Vergleich zu Placebo (Tabelle 5). Die Verbesserung des HbA1c im Vergleich zu Placebo war über alle Subgruppen hinweg generell konsistent, wobei die Subgruppen nach Geschlecht, Alter, Rasse, BMI-Ausgangswert, Zeitraum seit der Diabetesdiagnose, Vorhandensein des metabolischen Syndroms oder Standardindizes der Insulinresistenz (HOMA-IR) bzw. Insulinsekretion (HOMA-β) definiert waren. In keiner der Gruppen wurde eine bedeutende Änderung des Körpergewichts im Vergleich zum Ausgangswert beobachtet.
Tabelle 5:
Glykämische Parameter und Körpergewicht bei der abschliessenden Untersuchung (24-wöchige Studie) nach Einnahme von Sitagliptin als Add-on-Kombinationstherapie mit Metformin und einer stabilen Dosis Insulin †
Sitagliptin 100 mg | Placebo | |
HbA1c (%) | N = 223 | N = 229 |
Ausgangswert (Mittel) | 8.73 | 8.60 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel‡) | -0.66 | -0.13 |
Unterschied gegenüber Placebo (adjustiertes Mittel‡,§) | -0.53** | |
Patienten (%) mit HbA1c <7% | 32 (14.3) | 12 (5.2) |
Nüchtern-Glucose (mmol/l) | N = 225 | N = 229 |
Ausgangswert (Mittel) | 9.5 | 9.7 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel‡) | -1.2 | -0.2 |
Unterschied gegenüber Placebo (adjustiertes Mittel‡) | -1.0** | |
2-Stunden-PPG (mmol/l) | N = 182 | N = 189 |
Ausgangswert (Mittel) | 15.4 | 15.4 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel‡) | -2.1 | 0.1 |
Unterschied gegenüber Placebo (adjustiertes Mittel‡) | -2.2** | |
Körpergewicht (kg)¶ | N = 201 | N = 200 |
Ausgangswert (Mittel) | 87.9 | 88.0 |
Veränderung gegenüber Ausgangswert (adjustiertes Mittel‡) | -0.1 | 0.0 |
Unterschied gegenüber Placebo (adjustiertes Mittel‡) | 0.1# |
† «All-Patients-Treated» Population (Intention-to-treat-Analyse).
‡ Mittelwerte der kleinsten Quadrate, adjustiert für Insulin Behandlung bei Visite 1 (fertig hergestellt vs. nicht fertig hergestellt [intermediär- oder lang-wirksam]) und Ausgangswert.
§ Behandlung nach Insulin Stratum Interaktion war nicht signifikant (p>0.10).
** p<0.001 im Vergleich zu Placebo.
¶ «All-Patients-as-Treated» (APaT) Population unter Ausschluss von Daten nach einer glykämischen Notfallbehandlung.
# Statistisch nicht signifikant (p≥0.05) im Vergleich zu Placebo.
In einer anderen 24-wöchigen, randomisierten, doppelblinden und placebo-kontrollierten Studie zur Erfassung der Insulin-sparenden Wirkung von Sitagliptin als Add-on-Kombinationstherapie wurden 660 Patienten mit ungenügender glykämischer Kontrolle unter Insulin Glargin mit oder ohne Metformin (≥1500 mg pro Tag) zu entweder 100 mg Sitagliptin (N=330) oder Placebo (N=330) randomisiert. Die Verabreichung erfolgte einmal täglich während einer Intensivierung der Insulin-Therapie. Bei den Patienten mit Metformin war der HbA1c-Ausgangswert bei 8.70% und die Anfangsdosis von Insulin bei 37 IU/Tag. Patienten wurden instruiert ihre Insulin Glargin Dosis auf Grund der durch Fingerpunktion erhaltenen Nüchternblutzuckerwerte zu titrieren. Sekundäre Studien-Endpunkte beinhalteten HbA1c und FPG.
Bei den Patienten mit Metformin war zu Woche 24 der Anstieg der täglichen Insulindosis bei mit Sitagliptin behandelten Patienten geringer (19 IU/Tag, N=285) als bei Patienten unter Placebo (24 IU/Tag, N=283). Dieser Unterschied war statistisch signifikant (p=0.007).
Hinsichtlich sekundärer Studien-Endpunkte kam es zu einer HbA1c-Reduktion bei mit Sitagliptin, Metformin und Insulin behandelten Patienten von -1.35% verglichen mit -0.90% bei Patienten unter Placebo, Metformin, und Insulin, einer Differenz von -0.45% entsprechend [95% CI: -0.62, -0.29]. Die FPG-Reduktion bei mit Sitagliptin, Metformin und Insulin behandelten Patienten betrug ‑3.0 mmol/l verglichen mit -2.4 mmol/l bei Patienten unter Placebo, Metformin, und Insulin, einer Differenz von -0.7 mmol/l entsprechend [95% CI: -1.0, -0.3].
Glipizidkontrollierte Studie in Kombination mit Metformin mit nicht verzögerter Freisetzung
Die Langzweitwirkung wurde in einer 52-wöchigen, randomisierten, doppelblinden, Glipizidkontrollierten Studie bei Patienten mit Diabetes Typ 2 untersucht. Patienten mit ungenügender Blutzuckereinstellung unter Metformin (≥1500 mg/Tag) erhielten entweder täglich 100 mg Sitagliptin (N=588) oder Glipizid (N=584) während 52 Wochen. Patienten mit Glipizid erhielten als Initialdosis 5 mg/Tag und wurden dann innerhalb der folgenden 18 Wochen selektiv, ohne eine signifikante Hypoglykämie zu erzeugen, bis zu einer Ziel-FPG von <110 mg/dl auftitriert. Eine Maximaldosis von 20 mg/Tag war zur optimalen Blutzuckereinstellung zugelassen. Danach wurde die Glipizid-Dosis konstant gehalten. Die durchschnittliche Glipizid-Dosis nach der Titration betrug 10.3 mg.
Beide Behandlungen führten zu einer statistisch signifikanten Verbesserung der Blutzuckereinstellung im Vergleich zum Ausgangswert. Nach 52 Wochen betrug die Reduktion des HbA1c-Ausgangswertes 0.67% für 100 mg Sitagliptin täglich und 0.67% für Glipizid, was die Gleichwertigkeit der zwei Wirkstoffe bestätigte. Die Reduktion der FPG war 0.56 mmol/l für Sitagliptin und 0.42 mmol/l für Glipizid. In einer post-hoc Analyse hatten Patienten mit einem höheren HbA1c-Ausgangswert (≥9%) in beiden Gruppen eine grössere Reduktion des HbA1c-Ausgangswertes (Sitagliptin -1.68%; Glipizid -1.76%). Die Behandlung mit Sitagliptin führte zu einer Reduktion des Verhältnisses Proinsulin/Insulin, einem Marker für die Insulinsynthese und Insulinfreisetzung. Mit Glipizid verschlechterte sich das Verhältnis Proinsulin/Insulin. Die Häufigkeit von Hypoglykämien war in der Sitagliptin Gruppe mit 4,9% signifikant niedriger als in der Glipizid Gruppe (32.0%). Bei Patienten, die mit Sitagliptin behandelt wurden, nahm das Körpergewicht signifikant ab, während Patienten, denen Glipizid verabreicht wurde, eine signifikante Erhöhung des Körpergewichts aufwiesen (-1.5 kg vs. +1.1 kg).
Metforminhydrochlorid
Die prospektive, randomisierte Studie (UKPDS) hat den langfristigen Vorteil einer intensiven Blutzuckerkontrolle bei Diabetes Typ 2 bestätigt. Die Analyse der Resultate bei übergewichtigen Patienten, die mit Metformin behandelt wurden, nachdem eine Diät allein nicht geholfen hatte, zeigte:
- in der Metformin-Gruppe eine signifikante Reduktion des absoluten Risikos einer Komplikation im Zusammenhang mit Diabetes (29,8 Vorfälle/1000 Patientenjahre) versus Diät allein (43,3 Vorfälle/1000 Patientenjahre), p=0.0023, sowie versus die kombinierten Sulfonyl-harnstoff- und Insulin-Monotherapiegruppen (40.1 Vorfälle/1000 Patientenjahre), p=0.0034.
- eine signifikante Reduktion des absoluten Mortalitätsrisikos im Zusammenhang mit Diabetes: Metformin 7.5 Todesfälle/1000 Patientenjahre, Diät allein 12.7 Todesfälle/1000 Patientenjahre, p=0.017.
- eine signifikante Reduktion des absoluten Risikos der Gesamtmortalität: Metformin 13.5 Todesfälle/1000 Patientenjahre versus Diät allein 20.6 Todesfälle/1000 Patientenjahre (p=0.011), und versus die kombinierten Sulfonylharnstoff- und Insulin-Monotherapiegruppen 18.9 Todesfälle/1000 Patientenjahre (p=0.021).
- eine signifikante Reduktion des absoluten Risikos eines Herzinfarktes: Metformin 11 Vorfälle/1000 Patientenjahre, Diät allein 18 Todesfälle/1000 Patientenjahre (p=0.01).
TECOS kardiovaskuläre Sicherheitsstudie
Die TECOS-Studie (Trial Evaluating Cardiovascular Outcomes with Sitagliptin) war randomisiert und umfasste 14'671 Patienten in der Intention-to-treat Population mit HbA1c-Werten von ≥6.5 bis 8.0% und nachgewiesener kardiovaskulärer Erkrankung. Die Patienten erhielten entweder Sitagliptin 100 mg täglich (7332) (oder 50 mg täglich, falls der eGFR-Ausgangswert ≥30 und <50 ml/min/1,73 m2 war) oder Placebo (7339), gegeben zusätzlich zur üblichen Therapie mit Zielwerten gemäss regionalen Standards für HbA1c und kardiovaskulären Risikofaktoren. Patienten mit einer eGFR <30 ml/min/1.73 m2 wurden nicht in die Studie aufgenommen. Die Studie umfasste 2004 Patienten, die ≥75 Jahre alt waren und 3324 Patienten, die Niereninsuffizienz (eGFR <60 ml/min/1.73 m2) hatten.
Die Patienten in der Sitagliptin-Gruppe erhielten weniger antihyperglykämische Arzneimittel als jene in der Placebo-Gruppe (Hazard Ratio 0.72; 95% KI, 0.68-0.77; p≤0.001). Zudem war bei Patienten, die bei Studieneintritt nicht unter Insulin waren, das Starten einer chronischen Insulintherapie weniger wahrscheinlich (Hazard-Ratio 0.70; 95% KI, 0.63-0.79; p<0.001).
Der primäre kardiovaskuläre Endpunkt war eine Zusammensetzung des ersten Auftretens von kardiovaskulärem Tod, nicht-tödlichem Myokardinfarkt, nicht-tödlichem Schlaganfall oder Hospitalisierung wegen instabiler Angina pectoris. Sekundäre kardiovaskuläre Endpunkte waren erstes Auftreten von kardiovaskulärem Tod, nicht-tödlichem Myokardinfarkt oder nicht-tödlichem Schlaganfall; erstes Auftreten der einzelnen Komponenten der primären Zusammensetzung; Gesamtmortalität; und Hospitalisierungen aufgrund von Herzinsuffizienz.
Nach einer medianen Beobachtungszeit von drei Jahren erhöhte sich das Risiko für schwere unerwünschte kardiovaskuläre Ereignisse oder einer Hospitalisierung wegen Herzinsuffizienz nicht, wenn Sitagliptin bei Patienten mit Diabetes Typ 2 zusätzlich zur üblichen Therapie verabreicht wurde (Tabelle 6).
Tabelle 6:
Ergebnisraten für kardiovaskuläre Ereignisse und wichtige sekundäre Ereignisse
Sitagliptin 100 mg | Placebo | Hazard Ratio | p-Wert† | |||
N (%) | Inzidenz-Rate pro 100 Patientenjahre* | N (%) | Inzidenz-Rate pro 100 Patientenjahre | |||
Analyse der Intention-to-Treat Population | ||||||
Anzahl Patienten | 7332 | 7339 | 0.98 (0.89–1.08) | <0.001 | ||
Zusammengesetzter primärer Endpunkt (Kardiovaskulärer Tod, nicht-tödlicher Myokardinfarkt, nicht-tödlicher Schlaganfall, oder Hospitalisierung wegen instabiler Angina pectoris) | 839 (11.4) | 4.1 | 851 (11.6) | 4.2 | ||
Zusammengesetzter sekundärer Endpunkt (Kardiovaskulärer Tod, nicht-tödlicher Myokardinfarkt, oder nicht-tödlicher Schlaganfall) | 745 (10.2) | 3.6 | 746 (10.2) | 3.6 | 0.99 (0.89–1.10) | <0.001 |
Sekundäre Ereignisse | ||||||
Kardiovaskulärer Tod | 380 (5.2) | 1.7 | 366 (5.0) | 1.7 | 1.03 (0.89-1.19) | 0.711 |
Alle Myokardinfarkte (tödlich und nicht-tödlich) | 300 (4.1) | 1.4 | 316 (4.3) | 1.5 | 0.95 (0.81–1.11) | 0.487 |
Alle Schlaganfälle (tödlich und nicht-tödlich) | 178 (2.4) | 0.8 | 183 (2.5) | 0.9 | 0.97 (0.79–1.19) | 0.760 |
Hospitalisierung wegen instabiler Angina pectoris | 116 (1.6) | 0.5 | 129 (1.8) | 0.6 | 0.90 (0.70–1.16) | 0.419 |
Tod jeglicher Ursache | 547 (7.5) | 2.5 | 537 (7.3) | 2.5 | 1.01 (0.90–1.14) | 0.875 |
Hospitalisierung wegen Herzinsuffizienz‡ | 228 (3.1) | 1.1 | 229 (3.1) | 1.1 | 1.00 (0.83–1.20) | 0.983 |
* Inzidenz-Rate pro 100 Patientenjahre wurde berechnet als 100× (totale Anzahl Patienten mit ≥1 Ereignis während der entscheidenden Expositionsperiode pro Total der Patientenjahre der Beobachtung).
† Basierend auf dem Cox-Modell stratifiziert nach Region. Für zusammengesetzte Endpunkte entsprechen die p-Werte einem Nicht-Unterlegenheits-Test, mit dem Ziel zu zeigen, dass die Hazard Ratio kleiner ist als 1.3. Für alle anderen Endpunkte entsprechen die p-Werte einem Test der Differenzen der Hazard Rates.
‡ Die Analyse der Hospitalisierung wegen Herzinsuffizienz wurde angepasst für Vorgeschichte von Herzinsuffizienz bei Studieneintritt.
Pharmakokinetik
Die Resultate einer Studie bei gesunden Probanden zeigten, dass die Kombinationstabletten Janumet XR (Sitagliptinphosphat/Metforminhydrochlorid mit verzögerter Wirkstofffreisetzung) 50 mg/500 mg und 100 mg/1000 mg bioäquivalent sind zur gleichzeitigen Einnahme der entsprechenden Dosen Sitagliptinphosphat (Januvia) und Metforminhydrochlorid mit verzögerter Wirkstofffreisetzung als separate Tabletten.
Ebenso wurde Bioäquivalenz zwischen zwei Tabletten Janumet XR 50 mg/500 mg und einer Tablette Janumet XR 100 mg/1000 mg gezeigt.
In einer Crossover-Studie bei gesunden Probanden waren die AUC und die Cmax für Sitagliptin und die AUC für Metformin nach Verabreichung einer einzelnen Janumet XR 50 mg/500 mg Tablette (Testformulierung) und nach Verabreichung einer einzelnen Janumet (Sitagliptinphosphat/Metforminhydrochlorid mit nicht verzögerter Freisetzung) 50 mg/500 mg Tablette ähnlich. Nach Verabreichung einer einzelnen Janumet XR 50 mg/500 mg Tablette (Testformulierung) war der mittlere Cmax-Wert für Metformin 30% tiefer und der mediane Tmax-Wert 4 Stunden später, verglichen mit den entsprechenden Werten nach Verabreichung einer einzelnen Janumet 50 mg/500 mg Tablette. Dies ist konsistent mit den erwarteten Charakteristika der modifizierten Freisetzung für Metformin der Janumet XR Formulierung.
Nach Verabreichung von zwei Janumet XR 50 mg/1000 mg Tabletten einmal täglich mit dem Abendessen während 7 Tagen bei gesunden, erwachsenen Probanden erreichten Sitagliptin und Metformin den Steady-State-Zustand am Tag 4 bzw. 5. Die medianen Tmax-Werte für Sitagliptin und Metformin beim Steady-State-Zustand lagen bei ca. 3 bzw. 8 Stunden nach Einnahme. Die medianen Tmax-Werte für Sitagliptin und Metformin nach Verabreichung einer einzelnen Tablette Janumet lagen bei 3 bzw. 3.5 Stunden nach Einnahme.
Pharmakokinetische Daten, die Distribution, Metabolismus und Elimination von Metformin und Sitagliptin charakterisieren, stammen aus Studien mit der Verabreichung der einzelnen Komponenten von Metformin mit nicht verzögerter Wirkstofffreisetzung und Sitagliptin und sie sind auf Janumet XR übertragbar.
Absorption
Janumet XR
Nach Verabreichung von Janumet XR Tabletten mit einem Frühstück mit hohem Fettgehalt war die AUC für Sitagliptin unverändert. Die mittlere Cmax war um 17% erniedrigt, obschon die mediane Tmax im Vergleich zum nüchternen Zustand unverändert war. Nach Verabreichung von Janumet XR mit einem Frühstück mit hohem Fettgehalt war die AUC für Metformin um 62% erhöht, die Cmax für Metformin um 9% erniedrigt und die mediane Tmax für Metformin verzögerte sich um 2 Stunden im Vergleich zum nüchternen Zustand.
Sitagliptinphosphat
Die absolute Bioverfügbarkeit von Sitagliptin beträgt ca. 87%. Die gleichzeitige Verabreichung einer fettreichen Mahlzeit zusammen mit Sitagliptinphosphat hatte keinen Einfluss auf die Pharmakokinetik von Sitagliptin.
Distribution
Sitagliptinphosphat
Das durchschnittliche Verteilungsvolumen im Steady-State-Zustand nach einer einzelnen, intravenösen 100-mg Dosis Sitagliptin bei gesunden Probanden beträgt ca. 198 Liter. Die reversibel an Plasmaproteine gebundene Sitagliptin-Fraktion ist gering (38%).
Metforminhydrochlorid
Studien zur Verteilung von Metformin mit verzögerter Wirkstofffreisetzung wurden nicht durchgeführt. Das apparente Verteilungsvolumen (V/F) von Metformin nach Einnahme oraler Einzeldosen von 850 mg Metforminhydrochlorid mit nicht verzögerter Wirkstofffreisetzung war durchschnittlich 654 ± 358 l. Metformin wird im Gegensatz zu den Sulfonylharnstoffen, die zu mehr als 90% an Plasmaproteine gebunden sind, kaum an Plasmaproteine gebunden. Metformin verteilt sich in den Erythrozyten, vermutlich in Funktion der Zeit. Bei normalen klinischen Dosen und Dosierschemata mit den Metforminhydrochlorid-Tabletten werden die Steady-State-Plasmakonzentrationen von Metformin innerhalb von 24-48 Stunden erreicht und diese sind normalerweise <1 µg/ml. Bei kontrollierten klinischen Studien mit Metformin waren die maximalen Metformin-Plasmaspiegel selbst bei Maximaldosierungen nie höher als 5 µg/ml.
Metabolismus
Sitagliptinphosphat
Sitagliptin wird in erster Linie unverändert im Urin ausgeschieden und nur zu einem geringen Teil metabolisiert. Ca. 79% des Sitagliptin wird unverändert im Urin ausgeschieden.
Nach einer oral eingenommenen [14C]-Sitagliptin Dosis wurde ca. 16% der Radioaktivität in Form von Sitagliptin-Metaboliten ausgeschieden. Sechs Metaboliten wurden als Spuren detektiert und es wird nicht angenommen, dass diese zur Plasma-DPP-4-Inhibitor-Aktivität des Sitagliptin beitragen. Durch in vitro Studien konnte gezeigt werden, dass CYP3A4, mit Beteiligung von CYP2C8, das wichtigste Enzym für den begrenzten Metabolismus von Sitagliptin ist.
Metforminhydrochlorid
Studien mit intravenösen Einzeldosen an gesunden Probanden zeigen, dass Metformin unverändert im Urin ausgeschieden wird, in der Leber nicht metabolisiert (beim Menschen wurden keine Metaboliten gefunden) und biliär nicht ausgeschieden wird.
Elimination
Sitagliptinphosphat
Nach Gabe einer oralen [14C]-Sitagliptin Dosis an gesunde Probanden wurde annähernd 100% der verabreichten Radioaktivität im Faeces (13%) oder Urin (87%) innerhalb einer Woche ausgeschieden. Die apparente terminale Halbwertszeit t1/2 nach einer oral verabreichten 100 mg Dosis Sitagliptin betrug ca. 12.4 Stunden und die renale Clearance betrug ca. 350 ml/min.
Die Eliminierung von Sitagliptin geschieht hauptsächlich durch die Nieren und involviert aktive tubuläre Sekretion. Sitagliptin ist ein Substrat für den organischen Anionentransporter-3 des Menschen (hOAT-3), der bei der renalen Eliminierung von Sitagliptin beteiligt sein dürfte. Die klinische Relevanz von hOAT-3 für den Sitagliptin- Transport konnte noch nicht gezeigt werden. Sitagliptin ist ebenfalls ein Substrat für p-Glykoprotein, das auch bei der Vermittlung der renalen Eliminierung von Sitagliptin beteiligt sein könnte. Cyclosporin, ein p-Glykoprotein-Inhibitor, konnte jedoch die renale Clearance von Sitagliptin nicht reduzieren.
Metforminhydrochlorid
Die renale Clearance ist ca. 3.5-mal grösser als die Kreatinin-Clearance, was darauf schliessen lässt, dass die tubuläre Sekretion den wichtigsten Weg der Metformin-Eliminierung darstellt. Nach oraler Verabreichung von Metformin mit nicht verzögerter Wirkstofffreisetzung wird ca. 90% des absorbierten Arzneimittels innerhalb der ersten 24 Stunden via Niere ausgeschieden, wobei die Eliminierungshalbwertszeit im Plasma ca. 6.2 Stunden beträgt. Im Blut beträgt die Eliminierungshalbwertszeit ca. 17.6 Stunden, was darauf schliessen lässt, dass die Erythrozytenmasse ein Verteilungsraum sein könnte.
Nierenfunktionsstörung
Sitagliptinphosphat
Bei Patienten mit mittelschwerer Nierenfunktionsstörung mit eGFR von 30 bis <45 ml/min/1.73 m2 wurde eine ca. 2-fache Erhöhung der Plasma-AUC von Sitagliptin beobachtet, und bei Patienten mit schwerer Nierenfunktionsstörung (eGFR <30 ml/min/1.73 m2), einschliesslich Patienten mit terminaler Niereninsuffizienz (end-stage renal disease, ESRD) unter Hämodialyse, wurde eine ca. 4-fache Erhöhung im Vergleich zu Probanden mit normaler Nierenfunktion beobachtet.
Metforminhydrochlorid
Bei Patienten mit verminderter Nierenfunktion ist die Halbwertszeit von Metformin mit nicht verzögerter Wirkstofffreisetzung im Plasma und im Blut verlängert und die renale Clearance verringert (siehe KONTRAINDIKATIONEN und WARNHINWEISE UND VORSICHTSMASSNAHMEN).
Leberfunktionsstörung
Sitagliptinphosphat
Nach Verabreichung einer einzelnen 100 mg Dosis Sitagliptin stiegen bei Patienten mit mittelschwerer Leberfunktionsstörung (Child-Pugh Score 7 bis 9) die durchschnittlichen AUC und Cmax von Sitagliptin ca. 21% bzw. 13%, im Vergleich zur gesunden Kontrollgruppe. Diese Unterschiede werden als klinisch nicht von Bedeutung erachtet.
Es gibt keine klinischen Daten für Patienten mit schwerer Leberfunktionsstörung (Child-Pugh Score >9). Da jedoch Sitagliptin vor allem durch die Niere ausgeschieden wird, dürfte eine schwere Leberfunktionsstörung die Pharmakokinetik von Sitagliptin kaum beeinflussen.
Metforminhydrochlorid
Für Patienten mit Leberfunktionsstörung gibt es keine pharmakokinetischen Studien mit Metformin.
Anwendung bei älteren Personen
Sitagliptinphosphat
Gemäss einer populations-pharmakokinetischen Analyse von Phase I- und Phase II-Daten, hat das Alter keine klinisch signifikanten Auswirkungen auf die Pharmakokinetik von Sitagliptin. Ältere Personen (65 bis 80 Jahre) hatten im Vergleich zu jüngeren Personen ca. 19% höhere Sitagliptin-Plasmakonzentrationen.
Metforminhydrochlorid
Es existieren nur wenige Daten aus kontrollierten pharmakokinetischen Studien über Metformin mit nicht verzögerter Wirkstofffreisetzung bei gesunden älteren Personen. Diese deuten darauf hin, dass bei gesunden älteren Personen im Vergleich zu jungen gesunden Probanden die Gesamtplasma-Clearance von Metformin erniedrigt, die Halbwertszeit verlängert und Cmax erhöht ist.
Diese Daten lassen vermuten, dass die Veränderung der Pharmakokinetik von Metformin mit zunehmendem Alter primär auf die Veränderung der Nierenfunktion zurückzuführen ist (siehe GLUCOPHAGE1 Fachinformation).
1 GLUCOPHAGE® ist eine registrierte Marke der Merck Sante S.A.S, Partnergesellschaft der Merck KGaA in Darmstadt, Deutschland.
Verwendung bei Kindern und Jugendlichen
Die Sicherheit und Wirksamkeit von Janumet XR bei Kindern und Jugendlichen wurde nicht untersucht.
Geschlecht
Sitagliptinphosphat
Gemäss einer Analyse pharmakokinetischer Phase I-Daten und einer populationspharmakokinetischen Analyse von Phase I- und Phase II-Daten hat das Geschlecht keine klinisch signifikante Auswirkung auf die Pharmakokinetik von Sitagliptin.
Metforminhydrochlorid
Die pharmakokinetischen Parameter von Metformin von gesunden Probanden und Patienten mit Diabetes Typ 2 unterschieden sich nicht wesentlich, wenn sie entsprechend dem Geschlecht analysiert wurden. Desgleichen konnte in kontrollierten klinischen Studien bei Patienten mit Diabetes Typ 2 gezeigt werden, dass die antihyperglykämische Wirkung von Metformin bei Männern und Frauen ähnlich war.
Rasse
Sitagliptinphosphat
Gemäss einer Analyse pharmakokinetischer Phase I-Daten und einer populationspharmakokinetischen Analyse von Phase I- und Phase II-Daten, unter Einschluss von Weissen, Lateinamerikanern, Schwarzen, Asiaten und Angehörigen anderer Rassen, hat die Rasse keine klinisch signifikante Auswirkung auf die Pharmakokinetik von Sitagliptin.
Metforminhydrochlorid
Es existieren keine Studien über die pharmakokinetischen Parameter von Metformin entsprechend der Rasse. In kontrollierten klinischen Studien mit Metformin bei Patienten mit Diabetes Typ 2, erwies sich die antihyperglykämische Wirkung bei Weissen (n=249), Schwarzen (n=51) und Lateinamerikanern (n=24) als vergleichbar.
Body-Mass-Index (BMI)
Sitagliptinphosphat
Gemäss einer Analyse pharmakokinetischer Phase I-Daten und einer populationspharmakokinetischen Analyse von Phase I- und Phase II-Daten hat der Body-Mass-Index keine klinisch signifikante Auswirkung auf die Pharmakokinetik von Sitagliptin.
Präklinische Daten
Sitagliptin und Metformin
Präklinische toxikokinetische und orale Toxizitätsstudien mit Sitagliptin und Metformin als Kombinationspräparat mit nicht verzögerter Wirkstofffreisetzung wurden bei Hunden durchgeführt.
In einer 16-Wochen oralen Toxizitätsstudie wurde weiblichen Hunden 20 mg/kg/Tag Metformin allein oder in Kombination mit 2, 10 oder 50 mg/kg/Tag Sitagliptin verabreicht. Transiente Ataxie und/oder Tremor wurde in der Hochdosisgruppe beobachtet. Diese Anzeichen wurden als sitagliptinbedingt beurteilt, da sie in anderen Studien bei Hunden mit Sitagliptin allein bei 50 mg/kg/Tag beobachtet wurden. Der no-effect level für die behandlungsbedingten Veränderungen lag in dieser Studie bei 10 mg/kg/Tag Sitagliptin und 20 mg/kg/Tag Metformin, was etwa einer 6fachen systemischen Exposition einer Tagesdosis von 100 mg Sitagliptin sowie einer 2.5fachen systemischen Exposition einer Tagesdosis von 2000 mg Metformin beim Menschen entspricht.
Es wurden keine präklinischen Studien zur Kanzerogenität, Mutagenität, Beeinträchtigung der Fertilität oder Effekte auf die Reproduktion mit dem Kombinationsprodukt Janumet XR durchgeführt. Die folgenden Daten sind die Ergebnisse von separaten Studien mit Sitagliptin bzw. Metformin allein.
Sitagliptin
In nicht-klinischen Studien mit Ratten wurden Wirkungen von Sitagliptin nur bei Expositionen beobachtet, die weit über der Humandosis lagen, sodass sie für die normale Verwendung beim Menschen nicht in Betracht gezogen werden müssen. In präklinischen Sicherheitsstudien erwies sich Sitagliptin weder als genotoxisch noch als mutagen.
In einer Serie von Toxizitätsstudien mit wiederholter Gabe des Arzneimittels an Hunde, wurden Dosen von 2, 10 und 50 mg/kg/Tag während bis zu 53 Wochen untersucht. Nach 53 Wochen Behandlung mit einer Dosis von 10 mg/kg/Tag, was etwa dem 6-fachen der empfohlenen Dosis für den erwachsenen Menschen von 100 mg/Tag entspricht, wurden keine Effekte festgestellt. Bei Hunden, die mit 50 mg/kg/Tag Sitagliptin (ca. das 26-fache der Humanexposition) behandelt worden waren, traten im Zusammenhang mit der Therapie vorübergehend körperliche Symptome auf, beispielsweise Atmen mit geöffnetem Mund, Speichelfluss, weisslich- schaumartiges Erbrechen, Ataxia, Zittern, verringerte Aktivität und/oder gekrümmte Körperhaltung. Bei den 14- und 27-wöchigen Toxizitätsstudien mit einer Dosis von 50 mg/kg/Tag war zudem histologisch eine sehr schwache bis leichte Degeneration der Skelettmuskeln beobachtbar. In der 53-wöchigen Toxizitätsstudie wurde jedoch keine Degeneration der Skelettmuskeln mehr festgestellt, was darauf hindeutet, dass dies eine nicht reproduzierbare Wirkung war, oder dass diese Veränderung bei längerer Dauer der Behandlung nicht mehr auftrat. Die systemische Exposition im NOEL Bereich war in der 53-Wochenstudie (10 mg/kg/Tag) um den Faktor 5 höher als in der 27-Wochenstudie (2 mg/kg/Tag).
Weitere 3-monatige orale Toxizitätsstudien wurden bei Rhesus- und Cynomolgus-Affen durchgeführt. Die 3-Monatsstudie bei Rhesusaffen untersuchte das Potential von Sitagliptin für Hautläsionen oder Nierentoxizität, die Beurteilung beschränkte sich auf Haut und Niere. Die Tiere erhielten bis zu 100 mg Sitagliptin/kg/Tag (das 28fache der systemischen Exposition einer 100 mg Tagesdosis). Es gab keine behandlungsbedingten antemortem und postmortem Befunde.
In der 3-monatigen oralen Toxizitätsstudie bei Cynomolgus-Affen wurden die vollständigen Routineevaluationen durchgeführt. Die Tiere erhielten bis zu 100 mg Sitagliptin/kg/Tag (das 27fache der systemischen Exposition einer 100 mg Tagesdosis). Es gab keine behandlungsbedingten antemortem und postmortem Befunde.
Sitagliptin erwies sich bei Mäusen als nicht karzinogen, wenn 2 Jahre lang die maximal verträgliche Dosis von 500 mg/kg/Tag oral verabreicht wurde. Eine zweijährige Karzinogenitätsstudie wurde mit männlichen und weiblichen Ratten durchgeführt, denen orale Dosen von 50, 150, und 500 mg/kg/Tag Sitagliptin verabreicht wurde. Es wurde ein vermehrtes Auftreten von Leberzelladenomen und Karzinomen bei den männlichen Ratten mit hoher Dosis sowie von Leberkarzinomen bei den weiblichen Ratten mit hoher Dosis beobachtet. Basierend auf der empfohlenen täglichen Humandosis von 100 mg/Tag entsprach diese Dosis bei Ratten ca. dem 58-fachen der Humanexposition. Diese Dosis war bei den Rattenversuchen mit Hepatotoxizität assoziiert. Der No-observed-Effect-Level für die Induktion einer hepatischen Neoplasie betrug 150 mg/kg/Tag, das heisst, ca. das 19-fache der Humanexposition bei der empfohlenen 100 mg Dosis. Da gezeigt werden konnte, dass die Hepatotoxizität mit der Induktion einer hepatischen Neoplasie bei Ratten korreliert, ist anzunehmen, dass die grössere Häufigkeit von Lebertumoren bei Ratten die Folge der chronischen hepatischen Toxizität bei dieser hohen Dosis war. Die klinische Signifikanz dieses Befundes für den Menschen ist nicht bekannt.
Es wurden keine negativen Auswirkungen auf die Fruchtbarkeit von männlichen und weiblichen Ratten beobachtet, denen Sitagliptin vor oder während der Paarung oral mit Dosen von bis zu 1000 mg/kg/Tag verabreicht wurde (entsprechend ca. dem 100-fachen der Humanexposition, basierend auf der empfohlenen täglichen Dosis von 100 mg/Tag für den erwachsenen Menschen).
Reproduktionstoxikologische Studien an Ratten, die mit oralen Dosen von 1000 mg/kg/Tag behandelt wurden, ergaben im Zusammenhang mit der Behandlung eine leicht gestiegene Häufigkeit foetaler Rippenmissbildungen (fehlende Rippen, Hypoplasie und verbogene Rippen) in der Nachkommenschaft. Der No-observed-Effect-Level für Entwicklungsstörungen betrug 250 mg/kg/Tag (entsprechend dem 32fachen der Humanexposition, basierend auf der empfohlenen täglichen Dosis von 100 mg/Tag für den erwachsenen Menschen). Sitagliptin wird in der Milch laktierender Ratten ausgeschieden.
Es wurden keine Studien mit Sitagliptin, Metformin und Sulfonylharnstoff durchgeführt.
Metforminhydrochlorid
Präklinische Daten aus konventionellen Studien der Sicherheitspharmakologie, Toxizitätsstudien mit wiederholter Gabe des Arzneimittels sowie Studien der Genotoxizität, des karzinogenen Potenzials und der Reproduktionstoxizität zeigen, dass durch die Einnahme von Metformin keine speziellen Gefahren für Menschen entstehen.
Sonstige Hinweise
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung aufgedruckten Ablaufdatum «EXP» verwendet werden.
Besondere Lagerungshinweise
Das Arzneimittel muss ausserhalb der Reichweite von Kindern aufbewahrt werden.
Lagerung nicht über 30 °C. Die Flasche fest verschlossen halten, um den Inhalt vor Feuchtigkeit zu schützen.
Zulassungsnummer
65052 (Swissmedic).
Zulassungsinhaberin
MSD MERCK SHARP & DOHME AG, Luzern.
Stand der Information
September 2020.
HMV4+rhabdomyolysis/RCN000010607-CH+RCN000019721-CH
Composizione
Principi attivi
Sitagliptin fosfato monoidrato, metformina cloridrato.
Sostanze ausiliarie
Povidone K29-32, ipromellosa, idrossipropilcellulosa, silice colloidale anidra, sodio stearilfumarato (ciascuna compressa da 50 mg/500 mg contiene 1,26 mg di sodio; ciascuna compressa da 50 mg/1000 mg contiene 1,78 mg di sodio; ciascuna compressa da 100 mg/1000 mg contiene 1,77 mg di sodio), propile gallato (E310), macrogol 3350, caolino, titanio diossido (E171), cera carnauba e indigotina (E132).
Le compresse di Janumet XR 50 mg/500 mg contengono inoltre cellulosa microcristallina. Le compresse di Janumet XR 50 mg/1000 mg contengono inoltre ferro ossido giallo (E172).
Forma farmaceutica e quantità di principio attivo per unità
Compressa a rilascio modificato con 50 mg di sitagliptin come sitagliptin fosfato monoidrato e 500 mg di metformina cloridrato a rilascio prolungato (Janumet XR 50 mg/500 mg), 1000 mg di metformina cloridrato a rilascio prolungato (Janumet XR 50 mg/1000 mg), nonché 100 mg di sitagliptin come sitagliptin fosfato monoidrato e 1000 mg di metformina cloridrato a rilascio prolungato (Janumet XR 100 mg/1000 mg).
Indicazioni/Possibilità d'impiego
Janumet XR è indicato in aggiunta alla dieta e all'esercizio fisico per regolare la glicemia in pazienti affetti da diabete mellito di tipo 2 che non hanno un sufficiente controllo della glicemia né con metformina, né con sitagliptin in monoterapia oppure in pazienti già in trattamento con una combinazione di sitagliptin e metformina.
Janumet XR è indicato anche in combinazione con una sulfonilurea (come terapia combinata triplice) in aggiunta alla dieta e all'esercizio fisico in pazienti affetti da diabete mellito di tipo 2 che non hanno un sufficiente controllo della glicemia con una terapia combinata con due dei tre seguenti principi attivi: metformina, sitagliptin o una sulfonilurea.
Nel caso in cui non sia possibile ridurre sufficientemente la glicemia attraverso la dieta ed un maggiore esercizio fisico e l'insulina: in combinazione con insulina.
Posologia/Impiego
Aspetti generali
La posologia della terapia anti-iperglicemica con Janumet XR deve essere personalizzata sulla base della terapia corrente del paziente, dell'efficacia e della tollerabilità, non superando la dose giornaliera massima raccomandata di 100 mg di sitagliptin.
Janumet XR va assunto una volta al giorno con un pasto preferibilmente la sera.
La dose deve essere aumentata gradualmente per evitare eventuali disturbi gastrointestinali causati dalla metformina.
Le compresse non devono essere divise, spezzate, frantumate o masticate prima die essere ingerite per preservare le modalità di rilascio modificate.
La concentrazione plasmatica della metformina aumenta assumendo Janumet XR insieme agli alimenti. È stata segnalata l'incompleta dissoluzione di compresse di Janumet XR che sono state escrete nelle feci. Non è noto se questo materiale presente nelle feci contenga principio attivo. Il controllo glicemico va pertanto monitorato con particolare attenzione nei pazienti che segnalano ripetutamente la presenza nelle feci di residui delle compresse.
A causa delle possibili variazioni del controllo glicemico, le modifiche della terapia del diabete mellito di tipo 2 richiedono sempre cautela e un adeguato monitoraggio. Questo vale anche per il passaggio da compresse di metformina non a rilascio prolungato a compresse di metformina a rilascio prolungato, come ad es. Janumet XR.
Raccomandazioni posologiche
La dose iniziale di Janumet XR dovrebbe essere basata sulla terapia in corso.
Janumet XR si assume una volta al giorno con un pasto preferibilmente la sera. Janumet XR compresse è disponibile nei seguenti dosaggi:
50 mg di sitagliptin/500 mg di metformina cloridrato a rilascio prolungato
50 mg di sitagliptin/1000 mg di metformina cloridrato a rilascio prolungato
100 mg di sitagliptin/1000 mg di metformina cloridrato a rilascio prolungato
I pazienti che utilizzano le compresse da 50 mg di sitagliptin/500 mg di metformina a rilascio prolungato o le compresse da 50 mg di sitagliptin/1000 mg di metformina a rilascio prolungato dovrebbero assumere due compresse insieme una volta al giorno. La compressa da 100 mg di sitagliptin/1000 mg di metformina a rilascio prolungato va assunta singolarmente una volta al giorno.
Pazienti che non hanno un adeguato controllo della glicemia con la metformina in monoterapia
Per i pazienti che non hanno un adeguato controllo della glicemia con metformina in monoterapia, la dose iniziale raccomandata di Janumet XR è complessivamente di 100 mg di sitagliptin al giorno più la dose di metformina precedentemente assunta.
Pazienti che non hanno un adeguato controllo della glicemia con il sitagliptin in monoterapia
Per i pazienti che non hanno un adeguato controllo della glicemia con il sitagliptin in monoterapia, la dose iniziale raccomandata di Janumet XR è complessivamente di 100 mg di sitagliptin al giorno e 1000 mg di metformina cloridrato. Se necessario, la dose di metformina può essere titolata per regolare la glicemia. Dovrebbe essere considerato un aumento graduale della dose per evitare eventuali disturbi gastrointestinali causati dalla metformina. I pazienti che assumono una monoterapia con sitagliptin la cui dose è stata modificata a causa di un disturbo della funzionalità renale non dovrebbero passare a Janumet XR (vedere CONTROINDICAZIONI).
Pazienti che passano a Janumet XR da una terapia combinata con sitagliptin e metformina
Ai pazienti che passano a Janumet XR da una terapia combinata con sitagliptin e metformina può essere somministrata la stessa dose iniziale di sitagliptin e metformina sotto forma di preparato combinato Janumet XR.
Pazienti che non hanno un adeguato controllo della glicemia con una terapia combinata di due dei tre seguenti antidiabetici: sitagliptin, metformina o una sulfonilurea
La dose usuale iniziale di Janumet XR deve avere complessivamente 100 mg di sitagliptin al giorno. Nello stabilire la dose iniziale di metformina, si deve tener conto della glicemia attuale e, in caso di precedente assunzione di metformina, del relativo dosaggio somministrato finora. La dose deve essere aumentata gradualmente per evitare eventuali disturbi gastrointestinali causati dalla metformina. Nei pazienti che assumono attualmente una sulfonilurea o iniziano una terapia con una sulfonilurea è consigliabile un dosaggio inferiore della sulfonilurea per ridurre il rischio di ipoglicemia indotta dalla sulfonilurea (vedere AVVERTENZE E MISURE PRECAUZIONALI).
Pazienti che non hanno un adeguato controllo della glicemia con una terapia combinata di due dei tre seguenti antidiabetici: sitagliptin, metformina o insulina
La dose usuale iniziale di Janumet XR deve avere complessivamente 100 mg di sitagliptin al giorno. Nello stabilire la dose iniziale di metformina, si deve tener conto della glicemia attuale e, in caso di precedente assunzione di metformina, del relativo dosaggio somministrato finora. La dose deve essere aumentata gradualmente per evitare eventuali disturbi gastrointestinali causati dalla metformina. Nei pazienti trattati attualmente con insulina o che iniziano una terapia con insulina è consigliabile un dosaggio inferiore dell'insulina per ridurre il rischio di ipoglicemia (vedere AVVERTENZE E MISURE PRECAUZIONALI).
Non sono stati condotti studi specifici sulla sicurezza e l'efficacia di Janumet XR in pazienti precedentemente trattati con altri antidiabetici orali e poi passati a Janumet XR. Qualsiasi modifica della terapia del diabete mellito di tipo 2 richiede cautela e un monitoraggio accurato, poiché possono presentarsi variazioni del controllo della glicemia.
Impiego nei pazienti con disturbo della funzionalità renale
La funzionalità renale va valutata a intervalli regolari sia prima dell'inizio sia durante la terapia con Janumet XR.
Janumet XR è controindicato nei pazienti con un tasso presunto di filtrazione glomerulare (eGFR) <30 ml/min/1,73 m2. L'assunzione di Janumet XR deve essere sospesa nel caso in cui l'eGFR del paziente scenda al di sotto di 30 ml/min/1,73 m2 (vedere CONTROINDICAZIONI e AVVERTENZE E MISURE PRECAUZIONALI).
Non è consigliato l'inizio del trattamento con Janumet XR nei pazienti con un'eGFR ≥30 ml/min/1,73 m2 e <45 ml/min/1,73 m2. Nei pazienti che assumono Janumet XR e il cui eGFR successivamente scende al di sotto di 45 ml/min/1,73 m2 si devono valutare i benefici e i rischi connessi alla continuazione della terapia e si deve ridurre la dose di sitagliptin a 50 mg una volta al giorno.
Uso nei bambini e negli adolescenti
La sicurezza e l'efficacia di Janumet XR nei bambini e negli adolescenti non sono state esaminate.
Uso nei pazienti anziani
Poiché il sitagliptin e la metformina sono escreti principalmente per via renale, Janumet XR dovrebbe essere usato con cautela con l'aumentare dell'età del paziente. In particolar modo nei pazienti anziani è necessario il monitoraggio della funzionalità renale per prevenire l'acidosi lattica associata all'uso di metformina (vedere AVVERTENZE E MISURE PRECAUZIONALI, Metformina cloridrato, Acidosi lattica).
Controindicazioni
Janumet XR è controindicato nei pazienti con le condizioni seguenti.
- Grave disturbo della funzionalità renale (eGFR <30 ml/min/1,73 m2) (vedere AVVERTENZE E MISURE PRECAUZIONALI, Metformina cloridrato, Disturbo della funzionalità renale).
- Patologie acute che possono alterare la funzionalità renale, ad es. malattie cardiovascolari (compreso infarto miocardico), sepsi, disidratazione (diarrea, vomito ripetuto), gravi infezioni, ad es. delle vie urinarie, febbre alta.
- Ipersensibilità nota a sitagliptin fosfato, metformina cloridrato o a una qualsiasi delle sostanze ausiliarie di Janumet XR (vedere AVVERTENZE E MISURE PRECAUZIONALI, Reazioni di ipersensibilità ed EFFETTI INDESIDERATI, Esperienze post-marketing).
- Acidosi metabolica acuta o cronica, compresa chetoacidosi diabetica con o senza coma.
- La somministrazione endovascolare di agenti di contrasto iodati per esami radiografici può causare un'insufficienza renale e quindi un accumulo di metformina e acidosi lattica. Il trattamento con metformina deve essere interrotto 48 ore prima di un esame di questo tipo in caso di clearance della creatinina <60 ml/min ed eGFR <60 ml/min/1,73 m2. La terapia con metformina può essere ripresa solamente se da una verifica della funzione renale 48 ore dopo l'esame con agente di contrasto non è risultato alcun ulteriore peggioramento (vedere AVVERTENZE E MISURE PRECAUZIONALI).
Avvertenze e misure precauzionali
Janumet XR
Janumet XR non dovrebbe essere usato in pazienti affetti da diabete di tipo 1 o per il trattamento della chetoacidosi diabetica.
Pancreatite: tra i pazienti in terapia con sitagliptin sono stati osservati casi di pancreatite acuta, inclusi casi di pancreatite emorragica o necrotizzante fatale e non fatale (vedere EFFETTI INDESIDERATI). I pazienti dovrebbero essere informati sui sintomi caratteristici della pancreatite acuta: dolore addominale intenso e persistente. Un miglioramento della pancreatite è stato osservato dopo l'interruzione della terapia con sitagliptin. Qualora si sospetti la presenza di pancreatite, la terapia con Janumet XR e con altri medicamenti potenzialmente sospetti dovrebbe essere interrotta.
Controllo della funzionalità renale: sitagliptin e metformina sono escreti principalmente per via renale. Prima di iniziare una terapia con Janumet XR deve essere valutata la funzionalità renale, poiché il rischio di un accumulo di metformina e di acidosi lattica aumenta con la progressione dell'insufficienza renale. Janumet XR è controindicato nei pazienti con un grave disturbo della funzionalità renale (eGFR <30 ml/min/1,73 m2).
Durante il trattamento con Janumet XR, la funzionalità renale va valutata a intervalli regolari, come minimo:
- una volta all'anno nei pazienti con normale funzionalità renale;
- ogni 3-6 mesi nei pazienti con eGFR ≥45-59 ml/min/1,73 m2 e nei pazienti anziani;
- ogni 3 mesi nei pazienti con eGFR ≥30 44 ml/min/1,73 m2.
Janumet XR deve essere sospeso immediatamente nel caso in cui l'eGFR scenda al di sotto di 30 ml/min/1,73 m2.
In correlazione a medicamenti che possono causare ipoglicemia: è noto che gli antidiabetici come le sulfoniluree o l'insulina possono causare ipoglicemia. Pertanto, in caso di impiego di queste sostanze in combinazione con Janumet XR, può essere necessaria una riduzione della dose della sulfonilurea o dell'insulina.
Sitagliptin fosfato
Miopatia/Rabdomiolisi
In relazione all'uso di Janumet XR sono state segnalate miopatie, che si manifestano con dolore, sensibilità o debolezza muscolari associati a un forte aumento della creatinchinasi (CK, fino a 10 volte il limite superiore della norma).
La miopatia può talvolta presentarsi come rabdomiolisi con o senza insufficienza renale acuta secondaria a mioglobinuria, e i decessi sono stati rari.
I medici devono usare cautela nel prescrivere Janumet XR a pazienti con fattori predisponenti a una rabdomiolisi. Prima di iniziare il trattamento, un valore della creatinchinasi deve essere determinato nelle seguenti situazioni:
- compromissione della funzionalità renale
- ipotiroidismo non controllato
- anamnesi personale o familiare di disturbi muscolari ereditari
- anamnesi di tossicità muscolare con una statina o un fibrato
- dipendenza da alcol
- persone anziane (≥65 anni): in presenza di altri fattori predisponenti a una rabdomiolisi si deve prendere in considerazione la necessità di tale misurazione
- sesso femminile: in presenza di altri fattori predisponenti a una rabdomiolisi si deve prendere in considerazione la necessità di tale misurazione
In tali situazioni si deve prendere in considerazione il rischio di un trattamento rispetto al possibile beneficio.
In correlazione a medicamenti che possono causare ipoglicemia: in studi clinici con sitagliptin come monoterapia e parte integrante di una terapia combinata con principi attivi non associati a ipoglicemia (ad es. metformina o agonisti di PPARγ [tiazolidindioni]), la frequenza di un'ipoglicemia correlata all'assunzione di sitagliptin è stata all'incirca uguale a quella registrata nei pazienti che assumevano il placebo.
Reazioni di ipersensibilità
Nel corso dell'esperienza successiva all'immissione in commercio, sono state segnalate gravi reazioni di ipersensibilità in pazienti trattati con sitagliptin, uno dei principi attivi di Janumet XR. Queste reazioni comprendono anafilassi, angioedema e patologie cutanee esfoliative, inclusa la sindrome di Stevens-Johnson. Queste reazioni sono comparse nei 3 mesi successivi all'inizio della terapia con sitagliptin, in alcuni casi addirittura dopo la prima dose. Poiché questi eventi sono stati rilevati in una popolazione di grandezza sconosciuta, non è possibile fornire dati affidabili sulla loro frequenza. Nel caso in cui si sospetti una reazione di ipersensibilità, si dovrebbe interrompere la terapia con Janumet XR (vedere CONTROINDICAZIONI ed EFFETTI INDESIDERATI, Esperienze post-marketing).
Pemfigoide bolloso
Successivamente all'immissione in commercio, sono stati riportati casi di pemfigoide bolloso in pazienti trattati con inibitori della dipeptidil peptidasi-4 (DPP-4) che hanno reso necessaria un'ospedalizzazione. Nei casi segnalati i pazienti si sono in genere ripresi in seguito a trattamento immunosoppressivo topico o sistemico e dopo la sospensione dell'inibitore della DPP-4. Ai pazienti deve essere raccomandato di rivolgersi al proprio medico in caso di formazione di vescicole o erosioni cutanee nel corso del trattamento con Janumet XR. In caso di sospetto di pemfigoide bolloso, si dovrebbe interrompere la terapia con Janumet XR. Per la conferma della diagnosi e un adeguato trattamento, si dovrebbe considerare l'opportunità di indirizzare il paziente a un dermatologo.
Metformina cloridrato
Acidosi lattica: l'acidosi lattica è una complicanza metabolica rara, ma grave, che può insorgere in seguito a un accumulo di metformina nel corso del trattamento con Janumet XR (sitagliptin fosfato/metformina cloridrato a rilascio prolungato). L'acidosi lattica ha un decorso letale in circa il 50% dei casi. Può presentarsi anche in concomitanza con diversi stati patofisiologici, compreso il diabete mellito, nonché tutte le volte in cui si verificano ipoperfusione e ipossia tissutali significative. I segni tipici dell'acidosi lattica sono aumentati livelli di lattato nel sangue (>5 mmol/l), pH ematico ridotto, alterazioni elettrolitiche con aumentato gap anionico e aumentato rapporto lattato/piruvato. Se a causare l'acidosi lattica è la metformina, normalmente si rilevano livelli plasmatici di metformina >5 µg/ml.
La frequenza dell'acidosi lattica nei pazienti che assumono metformina cloridrato è molto bassa (circa 0,03 casi su 1000 anni paziente, con circa 0,015 decessi su 1000 anni paziente). In studi clinici con più di 20'000 anni paziente con esposizione alla metformina non sono stati riportati casi di acidosi lattica. I casi segnalati si sono verificati principalmente in pazienti diabetici con grave compromissione della funzionalità renale o insufficienza renale cronica. Nei pazienti con insufficienza cardiaca che necessitano un trattamento farmacologico, e in particolare nei pazienti con insufficienza cardiaca instabile o acuta con ipoperfusione e ipossia, sussiste un aumento del rischio di acidosi lattica. Il rischio di acidosi lattica aumenta con il grado di insufficienza renale e l'età del paziente. Il rischio può pertanto essere ridotto significativamente controllando regolarmente la funzione renale dei pazienti che assumono metformina e impiegando la dose minima efficace del medicamento. La funzione renale deve essere attentamente monitorata soprattutto nel corso del trattamento di pazienti anziani (vedere Uso nei pazienti anziani, Metformina cloridrato). La metformina non deve essere inoltre impiegata in nessuna condizione associata a ipossia, disidratazione e sepsi. Poiché un disturbo della funzionalità epatica può rallentare significativamente la degradazione del lattato, in generale si dovrebbe evitare la terapia con metformina in tutti i pazienti per i quali sulla base di esami clinici o di laboratorio sussiste il sospetto di una patologia epatica.
I pazienti che assumono metformina dovrebbero essere messi in guardia dall'eccessivo consumo di alcol, acuto o cronico, poiché l'alcol rafforza gli effetti della metformina cloridrato sul metabolismo del lattato. Inoltre, la somministrazione di metformina dovrebbe essere interrotta temporaneamente prima dell'iniezione intravasale di agenti di contrasto e prima di interventi chirurgici.
Spesso l'insorgenza di un'acidosi lattica è spesso subdola ed è accompagnata principalmente da sintomi aspecifici come ad esempio malessere, mialgie, dispnea, sonnolenza crescente e dolori addominali non specifici. In caso di acidosi più grave possono verificarsi ipotermia, ipotensione e bradiaritmia resistente. Il paziente e il medico curante devono essere consapevoli del possibile significato di questo tipo di sintomi e il paziente deve essere istruito ad informare immediatamente il medico se tali sintomi si manifestano. In un caso di questo tipo, la metformina va sospesa e il paziente deve essere ospedalizzato. I risultati di laboratorio di valore diagnostico sono i valori degli elettroliti e dei chetoni sierici, la glicemia, il valore del pH ematico e il livello di lattato e metformina.
Nei pazienti che assumono metformina, valori a digiuno di lattato plasmatico superiori ai valori normali, ma inferiori a 5 mmol/l, non indicano necessariamente un'imminente acidosi lattica e possono essere spiegati con altri meccanismi, come ad esempio un diabete non adeguatamente controllato o sovrappeso, un'attività fisica intensa o problemi tecnici nella gestione dei campioni.
L'acidosi lattica dovrebbe essere presa in considerazione in ogni paziente diabetico con acidosi metabolica, qualora non si rilevino segni di chetoacidosi (chetonuria e chetonemia).
L'acidosi lattica è un'emergenza medica che deve essere trattata in un ospedale. In un paziente affetto da acidosi lattica che assume metformina, il medicamento va sospeso immediatamente e vanno avviate le consuete misure di supporto. Poiché la metformina cloridrato e il lattato sono dializzabili (con una clearance fino a 170 ml/min in presenza di buone condizioni emodinamiche), si consiglia di sottoporre immediatamente il paziente a emodialisi. Questo trattamento consente spesso di ottenere una rapida eliminazione dei sintomi e un notevole recupero (vedere CONTROINDICAZIONI).
Una volta che il paziente è stabilizzato con la metformina, i sintomi gastrointestinali che spesso si presentano all'inizio della terapia scompaiono quasi del tutto. Se tali sintomi gastrointestinali dovessero ripresentarsi in un momento successivo, la causa potrebbe essere un'acidosi lattica o un'altra grave patologia.
Ipoglicemia: i pazienti che assumono metformina in monoterapia non presentano ipoglicemia in caso di uso normale. L'ipoglicemia correlata alla metformina può presentarsi in casi eccezionali, per es. in caso di apporto calorico insufficiente o forte consumo energetico (intenso sforzo fisico), nonché in correlazione all'assunzione contemporanea di alcol o di altre sostanze con effetto ipoglicemizzante (come ad esempio le sulfoniluree e l'insulina).
I pazienti anziani, deboli o malnutriti e quelli con insufficienza surrenale o ipofisaria o intossicazione da alcol, sono particolarmente soggetti agli effetti ipoglicemici. Talvolta l'ipoglicemia è difficile da riconoscere nei pazienti anziani e negli individui che assumono farmaci con azione bloccante dei recettori beta-adrenergici.
Assunzione contemporanea di altri medicamenti che possono influenzare la funzionalità renale o la disponibilità di metformina: i medicamenti concomitanti che possono influenzare la funzionalità renale, che causano una variazione significativa dell'emodinamica o influiscono sulla disponibilità di metformina, come ad esempio i medicamenti cationici escreti attraverso la secrezione tubulare renale (vedere INTERAZIONI, Metformina cloridrato), devono essere impiegati con cautela.
Somministrazione di agenti di contrasto iodati: la somministrazione intravascolare di agenti di contrasto iodati per esami radiografici può provocare un'insufficienza renale. Poiché questo può causare un accumulo di metformina e acidosi lattica, nei pazienti con eGFR <60 ml/min/1,73 m2 il trattamento con metformina va interrotto per tempo prima dell'esame (ca. 2 giorni) e può essere ripreso solamente se nei due giorni successivi all'esame con agente di contrasto la funzione renale non è peggiorata.
Ipossia: all'acidosi lattica sono stati associati arresto cardiocircolatorio di diverse origini, insufficienza cardiaca acuta ed infarto cardiaco acuto, così come altri stati in cui insorge ipossia. Questi stati possono causare anche un'azotemia prerenale. Se si dovessero verificare tali episodi nel corso della terapia con Janumet XR, il medicamento deve essere sospeso immediatamente.
Interventi chirurgici: Janumet XR deve essere interrotto temporaneamente in caso di interventi chirurgici (eccetto piccoli interventi che non limitano l'assunzione di cibo e bevande) e la terapia dovrebbe essere ripresa solo dopo il riavvio della nutrizione normale, sempre che la funzionalità renale sia stata valutata sufficiente (vedere POSOLOGIA/IMPIEGO).
Consumo di alcol: è noto che l'alcol rafforza l'effetto della metformina sul metabolismo del lattato. I pazienti che assumono Janumet XR devono perciò essere messi in guardia dall'eccessivo consumo di alcol acuto o cronico.
Sodio: questo medicamento contiene meno di 1 mmol (23 mg) di sodio per compressa, cioè essenzialmente «senza sodio».
Insufficienza epatica: poiché in alcuni casi l'insufficienza epatica è stata associata all'acidosi lattica, Janumet XR non dovrebbe essere utilizzato in pazienti per i quali sulla base di valutazioni cliniche o di laboratorio sussiste il sospetto di una patologia epatica.
Livelli ematici di vitamina B12: in studi clinici controllati con metformina per 29 settimane, in circa il 7% dei pazienti è stata osservata una riduzione a valori sotto la media dei livelli ematici di vitamina B12 in precedenza normali, ma senza manifestazioni cliniche. Una tale riduzione dei livelli ematici di vitamina B12, probabilmente dovuta a una riduzione dell'assorbimento della vitamina B12, è comunque associata molto raramente a un'anemia e sembra essere rapidamente reversibile subito dopo la sospensione della metformina o la somministrazione supplementare di vitamina B12. Si consiglia una valutazione annuale dei parametri ematici in tutti i pazienti che assumono Janumet XR. Tutte le alterazioni riscontrate devono essere esaminate accuratamente e trattate.
Le persone con inadeguato apporto o assorbimento di vitamina B12 o calcio sembrano essere predisposte a presentare ridotti livelli ematici di vitamina B12. Per questi pazienti è indicata una misurazione della concentrazione sierica di vitamina B12 da eseguirsi di routine ogni 2-3 anni.
Variazione dello stato clinico di pazienti affetti da diabete di tipo 2 precedentemente controllato: un paziente affetti da diabete di tipo 2 precedentemente ben controllato con Janumet XR, che però sviluppa improvvisamente alterazioni dei parametri di laboratorio o malattia clinica (malattie spesso vaghe e scarsamente definite) deve essere prontamente valutato per verificare l'eventuale presenza di chetoacidosi o acidosi lattica. Si devono valutare elettroliti e chetoni sierici e glicemia e, se indicato, pH del sangue e livelli ematici di lattato, piruvato e metformina. In caso di sospetta acidosi metabolica – indipendentemente dall'eziologia – si deve sospendere immediatamente Janumet XR e ospedalizzare il paziente.
Perdita del controllo glicemico: nei pazienti stabilizzati con un qualsiasi antidiabetico, in situazioni di stress come febbre, trauma, infezione o intervento chirurgico, può verificarsi una temporanea perdita di controllo della glicemia. In casi di questo tipo potrebbe essere necessario sospendere Janumet XR e sostituirlo temporaneamente con insulina. Al termine dell'episodio acuto si può riprendere la somministrazione di Janumet XR.
Impiego nei bambini e negli adolescenti
La sicurezza e l'efficacia di Janumet XR e di uno dei suoi principi attivi, sitagliptin, non sono state esaminate nei bambini e negli adolescenti di età inferiore ai 18 anni.
Impiego negli anziani
Janumet XR
Sitagliptin e metformina sono escreti principalmente per via renale. Pertanto, Janumet XR deve essere usato con cautela con l'aumentare dell'età del paziente. In particolar modo nei pazienti anziani è necessario il monitoraggio della funzionalità renale per prevenire l'acidosi lattica associata all'uso di metformina (vedere AVVERTENZE E MISURE PRECAUZIONALI, Metformina cloridrato, Acidosi lattica).
Sitagliptin fosfato
Negli studi clinici, la sicurezza e l'efficacia del sitagliptin nei pazienti anziani (≥65 anni) sono risultate simili a quelle rilevate nei pazienti più giovani (<65 anni).
Metformina cloridrato
Agli studi clinici controllati con metformina non ha partecipato un numero di pazienti anziani sufficientemente alto da poter affermare con sicurezza se essi reagiscano diversamente dai pazienti più giovani. Tuttavia, altre esperienze cliniche non evidenziano differenze tra pazienti più anziani e pazienti più giovani.
Janumet XR deve essere assunto di principio durante la cena. L'assunzione insieme agli alimenti può ridurre parzialmente gli effetti collaterali gastrointestinali della metformina. Inoltre, in caso di assunzione a stomaco vuoto è probabile una riduzione della biodisponibilità della metformina a rilascio prolungato. Questo potrebbe rendere più difficile il controllo del diabete. Si deve inoltre considerare che, contrariamente a quanto accade con la metformina a rilascio prolungato, la biodisponibilità della metformina non a rilascio prolungato viene ridotta dall'assunzione contemporanea di cibo.
Interazioni
Sitagliptin e metformina
In pazienti affetti da diabete di tipo 2, la combinazione di dosi multiple di sitagliptin (50 mg b.i.d.) e metformina (1000 mg b.i.d.) non ha alterato in modo rilevante la farmacocinetica del sitagliptin e della metformina.
Con Janumet XR non sono stati effettuati studi farmacocinetici di interazione. Tuttavia, tali studi sono stati condotti con i singoli principi attivi di Janumet XR, ossia con sitagliptin e metformina.
Sitagliptin fosfato
Negli studi farmacocinetici di interazione, il sitagliptin non ha evidenziato alcun influsso clinicamente significativo sulla farmacocinetica dei seguenti medicamenti: metformina, rosiglitazone, glibenclamide, simvastatina, warfarin e contraccettivi orali. In base a questi dati, il sitagliptin non inibisce gli isoenzimi del CYP CYP3A4, 2C8 e 2C9. Dati in vitro suggeriscono inoltre che il sitagliptin non inibisca CYP2D6, 1A2, 2C19 o 2B6 e non induca CYP3A4.
Nelle analisi farmacocinetiche di popolazione con pazienti affetti da diabete di tipo 2 non è risultato alcun influsso clinicamente significante delle terapie concomitanti sulla farmacocinetica del sitagliptin. Sono stati esaminati i medicamenti comunemente impiegati per il trattamento del diabete di tipo 2.
La somministrazione concomitante di sitagliptin e digossina ha determinato un lieve aumento dell'area sotto la curva (AUC, 11%) nonché del valore medio della concentrazione plasmatica massima (Cmax, 18%) della digossina. Queste variazioni non sono clinicamente significative. I pazienti che assumono digossina devono essere adeguatamente monitorati.
La somministrazione concomitante di una singola dose orale di sitagliptin da 100 mg e di una singola dose orale di ciclosporina da 600 mg, un potente inibitore della glicoproteina-P, ha aumentato la AUC e la Cmax del sitagliptin rispettivamente di circa il 29% e il 68%. Queste variazioni della farmacocinetica del sitagliptin non sono considerate clinicamente significative.
Metformina cloridrato
Glibenclamide: in uno studio di interazione con una singola somministrazione a pazienti affetti da diabete di tipo 2, la contemporanea assunzione di metformina e glibenclamide non ha determinato alcuna variazione della farmacocinetica e della farmacodinamica della metformina. Con la glibenclamide sono stati osservati ridotti valori della AUC e della Cmax, che però sono risultati molto variabili. Considerata la singola somministrazione del medicamento in questo studio e l'assenza di correlazione tra i livelli ematici di glibenclamide e l'effetto farmacodinamico, non è possibile esprimere alcun giudizio chiaro sulla significatività clinica di quest'interazione.
Furosemide: in uno studio di interazione con una singola somministrazione di metformina e furosemide a probandi sani è stato dimostrato un influsso dei parametri farmacocinetici di entrambi i medicamenti. La furosemide ha aumentato la Cmax plasmatica ed ematica della metformina del 22% e l'AUC della metformina nel sangue del 15%, senza influenzare significativamente la clearance renale della metformina. Rispetto alla monoterapia, la somministrazione concomitante di metformina ha determinato una riduzione della Cmax e dell'AUC della furosemide rispettivamente del 31% e del 12% e l'emivita terminale è risultata più breve del 32%, senza influenzare significativamente la clearance renale della furosemide. Non sono disponibili dati in merito all'interazione di metformina e furosemide in caso di uso prolungato.
Nifedipina: in uno studio di interazione con una singola somministrazione di metformina e nifedipina a soggetti volontari è stato dimostrato che la contemporanea somministrazione di nifedipina ha aumentato la Cmax e l'AUC plasmatica della metformina rispettivamente del 20% e del 9% e che questa è stata maggiormente escreta nelle urine. Il Tmax e l'emivita non sono stati influenzati. La nifedipina sembra accelerare l'assorbimento della metformina, mentre quest'ultima non ha influssi rilevanti sulla nifedipina.
L'uso concomitante di medicamenti che interferiscono con i sistemi di trasporto tubulare renale coinvolti nell'eliminazione renale della metformina (ad es. il trasportatore di cationi organici 2 [OCT2]/gli inibitori della proteina di estrusione multifarmaco e tossine [MATE] quali ranolazina, vandetanib, dolutegravir e cimetidina) potrebbe aumentare l'esposizione sistemica alla metformina e quindi il rischio di acidosi lattica. In studi di interazione con una somministrazione sia singola, sia multipla di metformina in combinazione con cimetidina a probandi sani, le concentrazioni massime di metformina nel plasma e nel sangue intero sono aumentate del 60%, mentre l'AUC plasmatica e l'AUC nel sangue intero della metformina sono aumentate del 40%. È necessario pertanto ponderare i benefici e i rischi dell'uso concomitante di queste sostanze con la metformina.
Altro: determinati medicamenti possono causare un'iperglicemia e compromettere quindi il controllo della glicemia. Tra questi rientrano ad es. tiazidici e altri diuretici, corticosteroidi, fenotiazine, medicamenti per la tiroide, estrogeni, contraccettivi orali, fenitoina, acido nicotinico, simpaticomimetici, calcio-antagonisti e isoniazide. In caso di somministrazione di uno di questi medicamenti a un paziente che assume Janumet XR, il paziente deve essere monitorato attentamente per garantire un adeguato controllo glicemico.
In studi di interazione con una singola somministrazione, nei probandi sani la farmacocinetica di metformina e propranololo e di metformina e ibuprofene non è risultata influenzata in caso di somministrazione combinata dei preparati.
La metformina si lega scarsamente alle proteine plasmatiche, per cui non interagisce quasi per niente con medicamenti fortemente legati alle proteine, come ad esempio salicilati, sulfonamidi, cloramfenicolo e probenecid, contrariamente alle sulfoniluree che invece si legano fortemente alle proteine sieriche.
Gravidanza/Allattamento
Utilizzo durante la gravidanza
Non sono stati condotti studi adeguati e rigorosamente controllati sull'uso di Janumet XR o dei suoi singoli componenti in donne in gravidanza.
Pertanto la sicurezza di Janumet XR nelle donne in gravidanza non è nota. L'assunzione di Janumet XR, nonché di altri antidiabetici orali, non è raccomandata in gravidanza.
Utilizzo durante l'allattamento
Con i principi attivi combinati di Janumet XR non sono stati condotti studi su animali in allattamento. Studi eseguiti con i singoli principi attivi hanno mostrato l'escrezione di metformina e sitagliptin nel latte di ratti che allattano. Non è noto se il sitagliptin venga escreto nel latte materno umano. Pertanto Janumet XR non deve essere assunto da donne che allattano.
Effetti sulla capacità di condurre veicoli e sull'impiego di macchine
Non sono stati condotti studi sugli effetti di Janumet XR sulla capacità di guidare veicoli e di utilizzare macchine. Si ritiene tuttavia che Janumet XR non alteri la capacità di guidare veicoli e di utilizzare macchine.
Effetti indesiderati
Non sono stati condotti studi clinici con Janumet XR compresse, ma è stato dimostrato che Janumet XR è bioequivalente a sitagliptin e metformina a rilascio prolungato somministrati in concomitanza (vedere FARMACOCINETICA).
In studi clinici controllati con placebo, la terapia combinata con sitagliptin e metformina non a rilascio prolungato è stata in generale ben tollerata dai pazienti affetti da diabete di tipo 2. L'incidenza complessiva di effetti indesiderati nei pazienti trattati con una terapia combinata con sitagliptin e metformina è stata simile a quella osservata nei soggetti che hanno assunto placebo e metformina.
Le frequenze sono definite come: comune ≥1/100, <1/10; non comune ≥1/1000, <1/100; raro ≥1/10'000, <1/1000; molto raro <1/10'000.
Terapia combinata con sitagliptin e metformina
In uno studio di 24 settimane, controllato con placebo, condotto su pazienti con controllo glicemico insufficiente tramite dieta ed esercizio fisico da soli, è stato valutato il trattamento con 50 mg di sitagliptin due volte al giorno in combinazione con 500 mg o 1000 mg di metformina due volte al giorno. Gli effetti indesiderati correlati alla terapia combinata che si sono verificati in ≥1% dei pazienti (e che sono stati più frequenti rispetto al gruppo di controllo con placebo) sono stati i seguenti.
Disturbi del metabolismo e della nutrizione
Comune: ipoglicemia.
Patologie del sistema nervoso
Comune: cefalea.
Disturbi gastrointestinali
Comune: diarrea, nausea, dispepsia, flatulenza, vomito.
In uno studio di 24 settimane, controllato con placebo, condotto con sitagliptin somministrato in aggiunta a una monoterapia con metformina già iniziata, 464 pazienti che assumevano metformina sono stati trattati con 100 mg di sitagliptin una volta al giorno, mentre altri 237 pazienti hanno ricevuto il placebo insieme alla metformina.
Gli effetti indesiderati correlati alla terapia combinata che si sono verificati in ≥1% dei pazienti (e che sono stati più frequenti rispetto al gruppo di controllo con placebo) sono stati i seguenti.
Patologie gastrointestinali
Comune: nausea.
Ipoglicemia e disturbi gastrointestinali
Negli studi controllati con placebo condotti con sitagliptin e metformina in terapia combinata, la frequenza di ipoglicemia (indipendentemente dalla valutazione della causalità da parte dello sperimentatore) in correlazione all'assunzione di sitagliptin in combinazione con metformina è stata all'incirca uguale a quella registrata nei pazienti che assumevano metformina in combinazione con placebo.
La frequenza dei disturbi gastrointestinali nei pazienti trattati con la combinazione di sitagliptin e metformina è stata paragonabile a quella dei pazienti trattati con metformina in monoterapia (vedere Tabella 1).
Tabella 1
Ipoglicemia e disturbi gastrointestinali (indipendentemente dalla valutazione della causalità da parte dello sperimentatore) osservati nei pazienti con la terapia combinata
Numero di pazienti (%) | ||||||
Studio condotto con sitagliptin e metformina su pazienti con un controllo glicemico insufficiente tramite la dieta e l'esercizio fisico | Studio con sitagliptin come terapia add-on con metformina | |||||
Placebo | Sitagliptin | Metformina | Sitagliptin | Placebo e metformina | Sitagliptin 100 mg q.d. e metformina | |
N = 176 | N = 179 | N = 364 | N = 372 | N = 237 | N = 464 | |
Ipoglicemia | 1 (0,6) | 1 (0,6) | 3 (0,8) | 6 (1,6) | 5 (2,1) | 6 (1,3) |
Diarrea | 7 (4,0) | 5 (2,8) | 28 (7,7) | 28 (7,5) | 6 (2,5) | 11 (2,4) |
Nausea | 2 (1,1) | 2 (1,1) | 20 (5,5) | 18 (4,8) | 2 (0,8) | 6 (1,3) |
Vomito | 1 (0,6) | 0 (0,0) | 2 (0,5) | 8 (2,1) | 2 (0,8) | 5 (1,1) |
Dolore addominale† | 4 (2,3) | 6 (3,4) | 14 (3,8) | 11 (3,0) | 9 (3,8) | 10 (2,2) |
† Nello studio condotto su pazienti con un controllo glicemico insufficiente tramite la dieta e l'esercizio fisico, i disturbi addominali sono stati conteggiati anche nei dolori addominali.
†† Dati aggregati per pazienti ai quali sono state somministrate dosi inferiori o superiori di metformina.
In tutti gli studi si è tenuto conto di tutti gli effetti collaterali designati come «ipoglicemia», ossia anche di quelli che non sono stati confermati tramite un esame dei livelli di glucosio nel sangue.
Sitagliptin in associazione con metformina e una sulfonilurea
In uno studio di 24 settimane, controllato con placebo, condotto con 100 mg di sitagliptin al giorno, somministrato in aggiunta a una terapia già iniziata con ≥4 mg al giorno di glimepiride e ≥1500 mg al giorno di metformina, gli effetti indesiderati correlati al medicamento osservati in ≥1% dei pazienti trattati con sitagliptin (N=116) e che si sono presentati con maggiore frequenza rispetto al gruppo di controllo con placebo (N=113) sono stati i seguenti.
Disturbi del metabolismo e della nutrizione
Comune: ipoglicemia.
Disturbi gastrointestinali
Comune: stipsi.
Nel corso del trattamento con sitagliptin in combinazione con metformina non sono state osservate variazioni clinicamente significative nelle funzioni vitali o nell'ECG (incluso l'intervallo QT).
Sitagliptin in combinazione con metformina e insulina
In uno studio di 24 settimane, controllato con placebo, condotto con 100 mg di sitagliptin somministrato in aggiunta a un trattamento combinato in corso con metformina ≥1500 mg al giorno e un dose stabile di insulina, l'ipoglicemia è stata l'unica reazione indesiderata osservata in ≥1% dei pazienti trattati con sitagliptin (N=229) la cui frequenza è stata superiore rispetto ai pazienti trattati con placebo (N=233) (sitagliptin 15,3%; placebo 8,2%). Questi dati di frequenza tengono conto di tutti gli effetti collaterali, indipendentemente dalla valutazione della causalità da parte dello sperimentatore. Soprattutto per i tassi di ipoglicemia si deve considerare che tutti i pazienti, oltre che con sitagliptin o placebo, sono stati trattati anche con insulina, in parte anche con metformina. In un altro studio di 24 settimane condotto su pazienti che nel corso di un'intensificazione della terapia con insulina (con o senza metformina) hanno ricevuto sitagliptin come terapia add-on, gli effetti indesiderati osservati in ≥1% dei pazienti trattati con sitagliptin e metformina (N=285) e la cui frequenza è stata superiore rispetto ai pazienti trattati con placebo e metformina (N=283), sono stati: stipsi (sitagliptin e metformina, 1,4%; placebo e metformina, 1,1%), diarrea (4,9%; 2,5%), vomito (3,2%; 1,1%), edema periferico (2,1%; 1,4%), febbre (1,1%; 0,4%), bronchite (1,4%; 1,1%), cellulite (1,4%; 1,1%), faringite (1,8%; 1,1%), infezione delle vie respiratorie superiori (4,2%; 1,4%), ridotta clearance della creatinina (1,1%; 0,0%), dolori del sistema muscoloscheletrico (1,4%; 1,1%), mialgia (1,1%; 0,7%), nefrolitiasi (1,1%; 0,4%) e tosse (2,8%; 1,8%). Questi dati di frequenza tengono conto di tutti gli effetti indesiderati, indipendentemente dalla valutazione della causalità da parte degli sperimentatori. L'incidenza dell'ipoglicemia è stata del 24,9% nei pazienti trattati con sitagliptin, metformina e insulina e del 37,8% in quelli trattati con placebo, metformina e insulina.
Effetti indesiderati noti con sitagliptin
Nei pazienti che assumevano sitagliptin non sono stati osservati effetti indesiderati correlati al medicamento con una frequenza ≥1%.
Effetti indesiderati noti con metformina
Patologie del sistema emolinfopoietico
Casi isolati di leucopenia, trombocitopenia e anemia emolitica.
Molto raro: ridotti livelli ematici di vitamina B12.
Disturbi del metabolismo e della nutrizione
Molto raro: acidosi lattica (incidenza 3 casi/100'000 anni paziente, vedere AVVERTENZE E MISURE PRECAUZIONALI).
Patologie del sistema nervoso
Comune: gusto metallico (3%).
Disturbi gastrointestinali
Comune: disturbi gastrointestinali come ad es. nausea, vomito, diarrea, dolore addominale, perdita di appetito.
Questi sintomi si presentano per lo più all'inizio della terapia e normalmente regrediscono spontaneamente.
Disturbi della funzionalità epatica e biliare
In singoli casi: valori anomali dei parametri di funzionalità epatica, ad es. aumento delle transaminasi o epatite (reversibile dopo la sospensione della metformina).
Patologie della cute e del tessuto sottocutaneo
Molto raro: reazioni cutanee come eritema, prurito, orticaria.
Studio sulla sicurezza cardiovascolare TECOS: lo studio TECOS (Trial Evaluating Cardiovascular Outcomes with Sitagliptin) ha coinvolto una popolazione intention-to-treat che comprendeva 7332 pazienti trattati con 100 mg di sitagliptin al giorno (o 50 mg al giorno se all'inizio dello studio il tasso presunto di filtrazione glomerulare [eGFR] era compreso tra ≥30 e <50 ml/min/1,73m2) e 7339 pazienti trattati con placebo. Entrambi i trattamenti sono stati aggiunti alla terapia solitamente utilizzata. La popolazione dello studio comprendeva complessivamente 2004 pazienti di età ≥75 anni (970 trattati con sitagliptin e 1034 con placebo). Nel complesso, l'incidenza globale di eventi indesiderati gravi nei pazienti trattati con sitagliptin è stata paragonabile a quella dei pazienti trattati con placebo. Una valutazione delle complicanze predefinite connesse al diabete ha evidenziato incidenze simili tra i gruppi, incluse le infezioni (18,4% dei pazienti trattati con sitagliptin e 17,7% di quelli trattati con placebo) e l'insufficienza renale (1,4% dei pazienti trattati con sitagliptin e 1,5% di quelli trattati con placebo). In generale, il profilo degli eventi indesiderati nei pazienti di età ≥75 anni è risultato simile a quello della popolazione complessiva.
Tra i pazienti che all'inizio dello studio stavano usando insulina e/o una sulfonilurea, l'incidenza di ipoglicemia grave è stata del 2,7% nei soggetti trattati con sitagliptin e del 2,5% in quelli trattati con placebo. Tra i pazienti che all'inizio dello studio non stavano usando insulina e/o una sulfonilurea, l'incidenza di ipoglicemia grave è stata dell'1,0% nei soggetti trattati con sitagliptin e dello 0,7% in quelli trattati con placebo. L'incidenza di pancreatite confermata da un comitato di verifica è stata dello 0,3% nei pazienti trattati con sitagliptin e dello 0,2% nei pazienti trattati con placebo. L'incidenza di neoplasie maligne confermate da un comitato di verifica è stata del 3,7% nei pazienti trattati con sitagliptin e del 4,0% nei pazienti trattati con placebo.
Pancreatite: in un'analisi aggregata di 19 studi clinici in doppio cieco condotti su 10'246 pazienti randomizzati al gruppo con 100 mg di sitagliptin al giorno (N = 5429) o con il relativo controllo (attivo o placebo) (N = 4817), l'incidenza di pancreatite acuta in ciascun gruppo è stata di 0,1 su 100 anni paziente (per il sitagliptin, 4 pazienti con un evento su 4708 anni paziente; per il controllo, 4 pazienti con un evento su 3942 anni paziente) (vedere anche Studio sulla sicurezza cardiovascolare TECOS e AVVERTENZE E MISURE PRECAUZIONALI, Pancreatite).
Esperienze post-marketing
Successivamente all'immissione in commercio di Janumet XR o sitagliptin, uno dei principi attivi di Janumet XR, sono state segnalate ulteriori reazioni indesiderate. Questi effetti indesiderati sono stati segnalati in caso di uso di Janumet XR o sitagliptin da soli e/o assieme ad altre sostante anti-iperglicemiche. Poiché essi si riferiscono a segnalazioni spontanee di una popolazione di grandezza sconosciuta, non è possibile stabilirne la frequenza o la causalità: reazioni di ipersensibilità, inclusi anafilassi, angioedema, eruzione cutanea, orticaria, vasculite cutanea e patologie cutanee esfoliative, inclusa la sindrome di Stevens-Johnson (vedere CONTROINDICAZIONI e AVVERTENZE E MISURE PRECAUZIONALI, Reazioni di ipersensibilità); pancreatite acuta, inclusa pancreatite emorragica con esito fatale e non fatale e pancreatite necrotizzante (vedere anche AVVERTENZE E MISURE PRECAUZIONALI); peggioramento della funzionalità renale, compresa insufficienza renale acuta (in determinati casi è necessaria la dialisi); rabdomiolisi; pemfigoide bolloso (vedere AVVERTENZE E MISURE PRECAUZIONALI, Pemfigoide bolloso); infezioni del tratto respiratorio superiore, rinofaringite; stipsi, vomito; cefalea; artralgia, mialgia, dolore agli arti, dolore dorsale; prurito.
Parametri di laboratorio
Sitagliptin fosfato
La frequenza delle variazioni dei parametri di laboratorio nei pazienti trattati con sitagliptin e metformina è stata simile a quella osservata nel gruppo di controllo trattato con placebo e metformina.
In diversi studi clinici è stato osservato un lieve aumento della conta leucocitaria (ca. 200 cellule/µl rispetto al placebo; valore basale medio della conta leucocitaria ca. 6600 cellule/µl) causato da un lieve aumento dei neutrofili. Quest'osservazione è stata fatta nella maggior parte degli studi, ma non in tutti. Queste variazioni dei parametri di laboratorio sono considerate clinicamente non rilevanti.
Metformina cloridrato
In studi clinici controllati con metformina per 29 settimane, in circa il 7% dei pazienti è stata osservata una diminuzione a valori sotto la media dei livelli ematici di vitamina B12 in precedenza normali, ma senza manifestazioni cliniche.
Una tale riduzione dei livelli ematici di vitamina B12, probabilmente dovuta a un'interferenza con l'assorbimento della vitamina B12 da parte del complesso fattore intrinseco-B12, è comunque associata molto raramente a un'anemia e sembra essere rapidamente reversibile subito dopo la sospensione della metformina o la somministrazione supplementare di vitamina B12 (vedere AVVERTENZE E MISURE PRECAUZIONALI, Metformina cloridrato).
La notifica di effetti collaterali sospetti dopo l'omologazione del medicamento è molto importante. Consente una sorveglianza continua del rapporto rischio-beneficio del medicamento. Chi esercita una professione sanitaria è invitato a segnalare qualsiasi effetto indesiderato sospetto, nuovo o serio, attraverso il portale online ElViS (Electronic Vigilance System). Maggiori informazioni sul sito www.swissmedic.ch.
Posologia eccessiva
Sitagliptin fosfato
Nell'ambito di studi clinici controllati condotti su probandi sani, sono state somministrate dosi singole di sitagliptin fino a 800 mg.
Con una dose di sitagliptin di 800 mg, in uno studio sono stati osservati aumenti minimi dell'intervallo QT, considerati però non clinicamente rilevanti.
Non vi sono dati di studi clinici condotti con dosi superiori a 800 mg.
In studi di fase I con somministrazione di dosi multiple di sitagliptin fino a 600 mg al giorno per periodi fino a 10 giorni e fino a 400 mg al giorno per periodi fino a 28 giorni non sono stati osservati effetti indesiderati dipendenti dalla dose clinicamente manifesti.
In caso di sovradosaggio, è opportuno fare uso delle comuni misure di supporto, per esempio rimuovere dal tratto gastrointestinale i principi attivi non ancora assorbiti, eseguire un monitoraggio clinico (incluso ECG) e istituire una terapia di supporto, qualora fosse necessario.
Il sitagliptin è moderatamente dializzabile. Negli studi clinici, circa il 13,5% della dose è stata rimossa nel corso di una sessione di emodialisi di 3-4 ore. Si può prendere in considerazione un'emodialisi più prolungata, se ritenuto appropriato dal punto di vista clinico. La dializzabilità del sitagliptin con dialisi peritoneale non è nota.
Metformina cloridrato
Si sono verificati sovradosaggi di metformina, anche superiori a 50 grammi. Nel 10% circa dei casi è stata osservata ipoglicemia, ma non è stato possibile dimostrare una correlazione causale con la metformina. Nel 32% circa dei casi di sovradosaggio con metformina è stata rilevata acidosi lattica (vedere AVVERTENZE E MISURE PRECAUZIONALI, Metformina cloridrato). Poiché la metformina è dializzabile (con una clearance fino a 170 ml/min in presenza di buone condizioni emodinamiche), si consiglia l'emodialisi per rimuovere la metformina accumulata.
Proprietà/Effetti
Codice ATC
A10BD07
Meccanismo d'azione
Il sitagliptin fosfato appartiene a una classe di antidiabetici orali, gli inibitori della dipeptidil peptidasi-4 (DPP-4), che migliorano il controllo glicemico nei pazienti affetti da diabete di tipo 2, aumentando le concentrazioni delle incretine attive. Le incretine, incluso il GLP-1 (Glucagon-like Peptide-1, peptide-1 glucagone-simile) e il GIP (Glucose-dependent insulinotropic Polypeptide, polipeptide insulinotropico glucosio-dipendente), vengono rilasciate dall'intestino nel corso dell'intera giornata e le loro concentrazioni vengono aumentate dall'assunzione di cibo. Le incretine fanno parte di un sistema endogeno coinvolto nella regolazione fisiologica dell'omeostasi del glucosio. Quando la glicemia è normale o elevata, GLP-1 e GIP aumentano la sintesi di insulina e il suo rilascio da parte delle cellule pancreatiche beta tramite segnali intracellulari con l'ausilio dell'AMP ciclico. Il trattamento del diabete di tipo 2 con GLP-1 o inibitori della DPP-4 in modelli animali ha evidenziato un miglioramento della sensibilità delle cellule beta al glucosio e una stimolazione della biosintesi e del rilascio di insulina. Maggiori concentrazioni di insulina aumentano la captazione del glucosio dei tessuti. Il GLP-1 diminuisce inoltre la secrezione di glucagone da parte delle cellule pancreatiche alfa. Minori concentrazioni di glucagone, insieme a maggiori concentrazioni di insulina, determinano una riduzione della produzione epatica di glucosio, che a sua volta si traduce in una riduzione della glicemia. L'effetto di GLP-1 e GIP è glucosio-dipendente. Quando la glicemia è bassa, il GLP-1 non stimola il rilascio di insulina né la soppressione della secrezione di glucagone. GLP-1 e GIP aumentano la secrezione di insulina se il glucosio supera i valori normali. Il GLP-1 non influisce sulla normale reazione del glucagone in caso di ipoglicemia. L'attività di GLP-1 e GIP è regolata dall'enzima DPP-4, che idrolizza rapidamente le incretine, trasformandole in sostanze inattive. Il sitagliptin impedisce l'idrolisi delle incretine da parte del DPP-4, per cui aumentano le concentrazioni plasmatiche delle forme attive di GLP-1 e GIP. Incrementando la concentrazione delle incretine attive, il sitagliptin stimola il rilascio di insulina e riduce le concentrazioni di glucagone in modo glucosio-dipendente. Nei pazienti affetti da diabete di tipo 2 con iperglicemia, queste variazioni delle concentrazioni di insulina e glucagone fanno ridurre la concentrazione di emoglobina A1c (HbA1c) e i livelli di glicemia postprandiale e a digiuno. Questo meccanismo glucosio-dipendente differisce da quello delle sulfoniluree, che inducono il rilascio di insulina anche in presenza di bassi valori di glucosio, comportando la possibilità di causare ipoglicemia nei pazienti affetti da diabete di tipo 2 e nei soggetti sani. Il sitagliptin è un inibitore potente e altamente selettivo dell'enzima DPP-4, e a concentrazioni terapeutiche non inibisce l'attività degli enzimi strettamente correlati DPP-8 o DPP-9.
Metformina cloridrato
La metformina è un antidiabetico che migliora la tolleranza al glucosio nei pazienti affetti da diabete di tipo 2 e abbassa sia i livelli basali che quelli postprandiali del glucosio plasmatico. I meccanismi d'azione farmacologici della metformina differiscono da quelli di altre classi di antidiabetici orali. La metformina riduce la sintesi di glucosio a livello epatico e l'assorbimento intestinale di glucosio, nonché migliora la sensibilità all'insulina, aumentando la captazione periferica di glucosio e la sua utilizzazione. A differenza delle sulfoniluree, la metformina non produce ipoglicemia né nei pazienti affetti da diabete di tipo 2, né nei probandi sani, eccetto in casi molto particolari (vedere AVVERTENZE E MISURE PRECAUZIONALI, Metformina cloridrato) e non causa iperinsulinemia. Nel corso di una terapia con metformina, la secrezione di insulina rimane invariata, mentre il livello di insulina a digiuno e la risposta dell'insulina plasmatica possono addirittura diminuire nel corso della giornata.
Farmacodinamica
Non pertinente.
Efficacia clinica
Studi clinici
Non sono stati condotti studi clinici per valutare l'efficacia e la sicurezza di Janumet XR. Studi clinici con somministrazione concomitante di sitagliptin e metformina non a rilascio prolungato hanno evidenziato miglioramenti significativi del controllo glicemico nei pazienti affetti da diabete di tipo 2. È stata dimostrata, per tutti i dosaggi, la bioequivalenza tra Janumet XR e la combinazione di sitagliptin e metformina compresse a rilascio prolungato.
In uno studio comparativo, la somministrazione una volta al giorno di metformina a rilascio prolungato è stata similmente efficace per tutti i parametri di misura per il controllo glicemico, come la somministrazione due volte al giorno di metformina non a rilascio prolungato normalmente prescritta.
Sitagliptin e metformina non a rilascio prolungato nei pazienti affetti da diabete di tipo 2
A uno studio di 24 settimane randomizzato, in doppio cieco e controllato con placebo, condotto per valutare la sicurezza e l'efficacia della combinazione di sitagliptin e metformina, hanno partecipato complessivamente 1091 pazienti affetti da diabete di tipo 2 con livelli glicemici non adeguatamente controllati nonostante la dieta e l'esercizio fisico.
Nei pazienti con un controllo glicemico insufficiente tramite la dieta e l'esercizio fisico da soli, la combinazione di sitagliptin e metformina ha prodotto miglioramenti significativi di HbA1c, FPG (glucosio plasmatico a digiuno) e PPG (glucosio plasmatico postprandiale) a 2 ore rispetto al placebo, alla monoterapia con metformina e alla monoterapia con sitagliptin (p<0,001; Tabella 2, Grafico 1). Un miglioramento con riduzione quasi massima del FPG è stato raggiunto nel primo endpoint dopo 3 settimane (conformemente al primo endpoint dopo l'inizio della terapia) e ha potuto essere mantenuto per tutte e 24 le settimane dello studio. Rispetto al placebo, i pazienti con un valore iniziale dell'HbA1c più elevato hanno presentato una maggiore riduzione media dell'HbA1c.
Il miglioramento dei valori di HbA1c non è stato influenzato da sesso, età, etnia o indice di massa corporea (BMI) iniziale. Anche misurazioni della funzione delle cellule beta (HOMA-β) e del rapporto proinsulina/insulina hanno evidenziato un miglioramento in caso di somministrazione di sitagliptin e metformina in terapia combinata rispetto alle rispettive monoterapie. Gli endpoint lipidici non sono stati influenzati. Le riduzioni medie dell'HbA1c per il sottogruppo di pazienti senza antidiabetici all'entrata nello studio sono state: sitagliptin 100 mg una volta al giorno (n=88), -1,06%; metformina 500 mg due volte al giorno (n=90), -1,09%; metformina 1000 mg due volte al giorno (n=87), -1,24%; sitagliptin 50 mg due volte al giorno con metformina 500 mg due volte al giorno (n=100), -1,59%; sitagliptin 50 mg due volte al giorno con metformina 1000 mg due volte al giorno (n=86), -1,94% e nei pazienti che hanno ricevuto placebo (n=83), -0,17%.
Grafico 1: variazione media dell'HbA1c rispetto al valore iniziale nel corso di 24 settimane con sitagliptin e metformina da soli e in combinazione nei pazienti affetti da diabete di tipo 2†
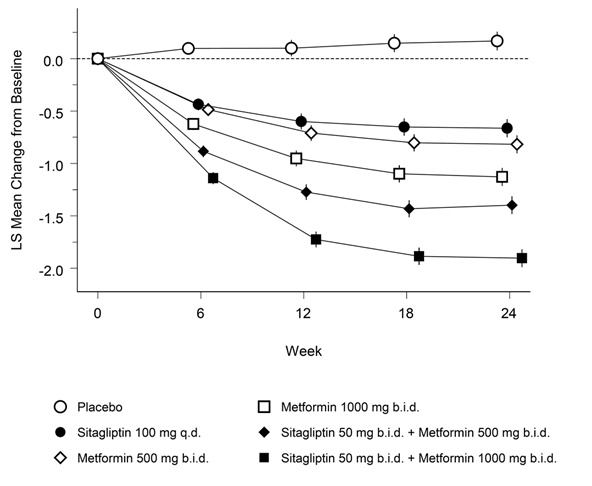
† All Patients Treated Population (analisi intention-to-treat), medie dei minimi quadrati aggiustate per stato della precedente terapia antidiabetica e valore iniziale.
Tabella 2
Parametri glicemici e peso corporeo rilevati nell'esame conclusivo dopo 24 settimane†
Placebo | Sitagliptin | Metformina | Sitagliptin | Metformina | Sitagliptin | |
HbA1c (%) | N = 165 | N = 175 | N = 178 | N = 183 | N = 177 | N = 178 |
Valore iniziale (medio) | 8,68 | 8,87 | 8,90 | 8,79 | 8,68 | 8,76 |
Variazione rispetto al valore iniziale (media aggiustata‡) | 0,17 | -0,66 | -0,82 | -1,40 | -1,13 | -1,90 |
Variazione rispetto al placebo (media aggiustata‡) | - | -0,83§ | -0,99§ | -1,57§ | -1,30§ | -2,07§ |
Pazienti (%) con HbA1c <7% | 15 (9,1) | 35 (20,0) | 41 (23,0) | 79 (43,2) | 68 (38,4) | 118 (66,3) |
FPG (mmol/l) | N = 169 | N = 178 | N = 179 | N = 183 | N = 179 | N = 180 |
Valore iniziale (medio) | 10,91 | 11,19 | 11,40 | 11,33 | 10,94 | 10,93 |
Variazione rispetto al valore iniziale (media aggiustata‡) | 0,32 | -0,97 | -1,52 | -2,62 | -1,63 | -3,55 |
Variazione rispetto al placebo (media aggiustata‡) | - | -1,29§ | -1,84§ | -2,94§ | -1,95§ | -3,87§ |
PPG a 2 ore (mmol/l) | N = 129 | N = 136 | N = 141 | N = 147 | N = 138 | N = 152 |
Valore iniziale (medio) | 15,38 | 15,86 | 16,26 | 16,21 | 15,74 | 15,94 |
Variazione rispetto al valore iniziale (media aggiustata‡) | 0,02 | -2,88 | -2,97 | -5,14 | -4,33 | -6,48 |
Variazione rispetto al placebo | -2,9§ | -2,98§ | -5,16§ | -4,35§ | -6,49§ | |
Peso corporeo (kg)** | N = 167 | N = 175 | N = 179 | N = 184 | N = 175 | N = 178 |
Valore iniziale (medio) | 90,1 | 85,9 | 88,1 | 90,0 | 89,4 | 88,2 |
Variazione rispetto al valore iniziale (media aggiustata‡) | -0,9 | 0,0 | -0,9 | -0,6 | -1,1 | -1,3 |
Variazione rispetto al placebo (media aggiustata‡) | 0,9. | 0,1# | 0,4# | -0,1# | -0,3# |
† All Patients Treated Population (analisi intention-to-treat).
‡ Medie dei minimi quadrati aggiustate per stato della precedente terapia antidiabetica e valore iniziale.
§ p<0,001 rispetto al placebo.
** All Patients as Treated (APaT) Population, ad esclusione dei dati dopo un trattamento glicemico d'urgenza.
¶ p=0,005 rispetto al placebo.
# Statisticamente non significativo (p≥0,05) rispetto al placebo.
Inoltre, questo studio includeva pazienti (N=117) con una forte iperglicemia (HbA1c >11% o glucosio ematico >15,56 mmol/l), trattati due volte al giorno con 50 mg di sitagliptin e 1000 mg di metformina. Per questi pazienti non c'è stato un controllo tramite placebo. In questo gruppo di pazienti il valore iniziale medio dell'HbA1c era 11,15%, l'FPG era 17,47 mmol/l e il PPG a 2 ore ammontava a 24,5 mmol/l. Dopo 24 settimane sono state rilevate le seguenti riduzioni rispetto al valore iniziale: ‑2,9% per HbA1c, ‑7,04 mmol/l per FPG e ‑11,55 mmol/l per il PPG a 2 ore. Dopo 24 settimane, in questa coorte è stato registrato un aumento del peso corporeo pari a 1,3 kg.
Sitagliptin come terapia add-on in pazienti con un controllo inadeguato della glicemia con la metformina in monoterapia
In due studi clinici in doppio cieco controllati con placebo sono state valutate la sicurezza e l'efficacia della combinazione di sitagliptin e metformina non a rilascio prolungato in pazienti affetti da diabete mellito di tipo 2. In entrambi gli studi sono stati randomizzati pazienti con livelli glicemici non adeguatamente controllati con dosi costanti di metformina ≥1500 mg e sono stati somministrati loro una volta al giorno 100 mg di sitagliptin o placebo in aggiunta alla terapia in corso con metformina.
In uno studio, a 701 pazienti sono stati somministrati una volta al giorno per 24 settimane 100 mg di sitagliptin o placebo. L'aggiunta di sitagliptin alla terapia in corso con metformina non a rilascio prolungato ha prodotto miglioramenti significativi di HbA1c (-0,65%), FPG (-1,41 mmol/l) e PPG a 2 ore (-2,81 mmol/l) rispetto all'aggiunta di placebo (vedere Grafico 2 e Tabella 3). Il miglioramento del valore di HbA1C rispetto al placebo non è stato influenzato da valore iniziale dell'HbA1C, precedenti terapie con antidiabetici, sesso, età, indice di massa corporea (BMI) iniziale, tempo trascorso dalla diagnosi di diabete, presenza di sindrome metabolica, indizi standard di insulino-resistenza (HOMA-IR) o secrezione di insulina (HOMA-β).
Grafico 2: variazione media del valore iniziale dell'HbA1c nel corso di 24 settimane con una dose di 100 mg al giorno di sitagliptin e metformina o placebo e metformina in pazienti affetti da diabete di tipo 2† ‡
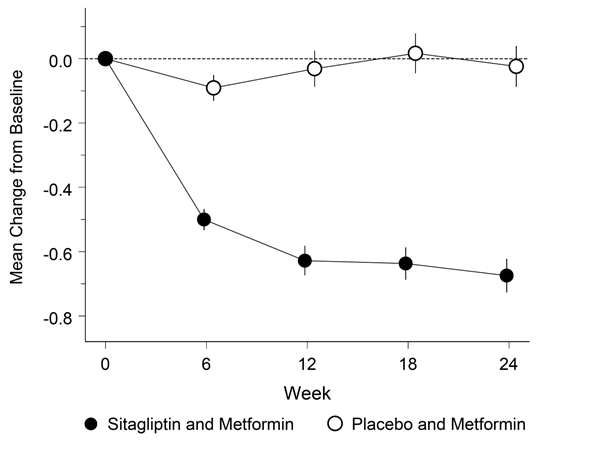
† Pazienti con metformina in monoterapia e inadeguato controllo della glicemia.
‡ All Patients Treated Population. Medie dei minimi quadrati aggiustate per stato della precedente terapia antidiabetica e valore iniziale.
Tabella 3
Parametri glicemici e peso corporeo rilevati nell'esame conclusivo (studio di 24 settimane)
Sitagliptin come terapia add-on- in pazienti con inadeguato controllo della glicemia in trattamento con metformina†
Sitagliptin 100 mg q.d. + metformina | Placebo + | |
HbA1c (%) | N = 453 | N = 224 |
Valore iniziale (medio) | 7,96 | 8,03 |
Variazione rispetto al valore iniziale (media aggiustata‡) | -0,67 | -0,02 |
Variazione rispetto a placebo + metformina (media aggiustata‡) | -0,65§ | |
Pazienti (%) con HbA1c <7% | 213 (47,0) | 41 (18,3) |
FPG (mmol/l) | N = 454 | N = 226 |
Valore iniziale (medio) | 9,44 | 9,64 |
Variazione rispetto al valore iniziale (media aggiustata‡) | -0,94 | 0,47 |
Variazione rispetto a placebo + metformina (media aggiustata‡) | -1,41§ | |
PPG a 2 ore (mmol/l) | N = 387 | N = 182 |
Valore iniziale (medio) | 15,25 | 15,13 |
Variazione rispetto al valore iniziale (media aggiustata‡) | -3,44 | -0,63 |
Variazione rispetto a placebo + metformina (media aggiustata‡) | -2,81§ | |
Peso corporeo (kg)** | N = 399 | N = 169 |
Valore iniziale (medio) | 86,9 | 87,6 |
Variazione rispetto al valore iniziale (media aggiustata‡) | -0,7 | -0,6 |
Variazione rispetto a placebo + metformina (media aggiustata‡) | -0,1¶ |
† All Patients Treated Population (analisi intention-to-treat).
‡ Medie dei minimi quadrati aggiustate per stato della precedente terapia antidiabetica e valore iniziale.
§ p<0,001 rispetto a placebo + metformina.
** All Patients as Treated (APaT) Population, ad esclusione dei dati dopo un trattamento glicemico d'urgenza.
¶ Statisticamente non rilevante (p≥0,05) rispetto a placebo + metformina.
Grafico 3: profili del glucosio plasmatico nelle 24 ore in pazienti affetti da diabete di tipo 2 dopo un trattamento di 4 settimane con sitagliptin 50 mg b.i.d. e metformina o placebo e metformina†
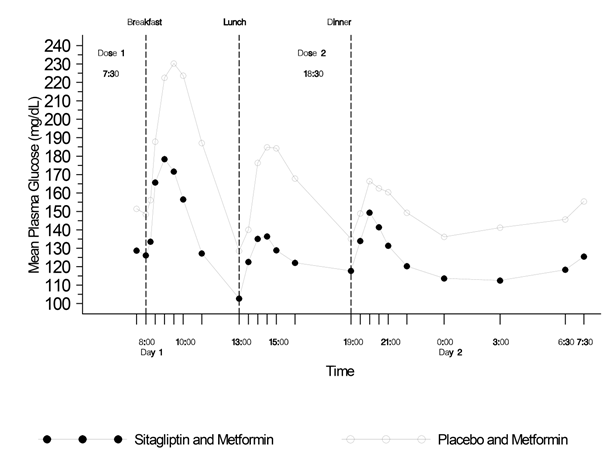
† Pazienti con inadeguato controllo della glicemia con la metformina in monoterapia.
Sitagliptin come terapia add-on in pazienti con inadeguato controllo della glicemia con una combinazione di metformina non a rilascio prolungato e glimepiride
Complessivamente 441 pazienti affetti da diabete di tipo 2 hanno partecipato per 24 settimane a uno studio randomizzato, in doppio cieco e controllato con placebo, condotto per valutare l'efficacia di 100 mg di sitagliptin una volta al giorno rispetto a placebo, in combinazione con glimepiride (come monoterapia o in combinazione con metformina). In questo studio 220 pazienti ricevevano già una terapia combinata con glimepiride (≥4 mg al giorno) e metformina (≥1500 mg al giorno).
La combinazione di sitagliptin, glimepiride e metformina ha prodotto una significativa riduzione del valore iniziale dell'HbA1c (-0,89%) e dell'FPG (-1,15 mmol/l) rispetto al placebo (vedere Tabella 4). Rispetto al placebo, i pazienti con un valore iniziale dell'HbA1c più elevato hanno presentato una maggiore riduzione media dell'HbA1c.
Tabella 4
Parametri glicemici* e peso corporeo rilevati nell'esame conclusivo (studio di 24 settimane) con sitagliptin in aggiunta alla terapia combinata con glimepiride e metformina†
Sitagliptin 100 mg | Placebo | |
HbA1c (%) | N = 115 | N = 105 |
Valore iniziale (medio) | 8,27 | 8,28 |
Variazione rispetto al valore iniziale (media aggiustata)‡ | -0,59 | 0,30 |
Variazione rispetto al placebo (media aggiustata‡) | -0,89§ | |
Pazienti (%) con HbA1c <7% | 26 (22,6) | 1 (1,0) |
FPG (mmol/l) | N = 115 | N = 109 |
Valore iniziale (medio) | 9,96 | 9,94 |
Variazione rispetto al valore iniziale (media aggiustata)‡ | -0,43 | 0,72 |
Variazione rispetto al placebo (media aggiustata‡) | -1,15§ | |
Peso corporeo (kg)** | N = 102 | N = 74 |
Valore iniziale (medio) | 86,5 | 84,6 |
Variazione rispetto al valore iniziale (media aggiustata)‡ | 0,4 | -0,7 |
Variazione rispetto al placebo (media aggiustata‡) | 1,1†† |
* I parametri glicemici primari e secondari erano HbA1c e glucosio a digiuno.
† All Patients Treated Population (analisi intention-to-treat).
‡ Medie dei minimi quadrati aggiustate per stato della precedente terapia antidiabetica e valore iniziale.
§ p<0,001 rispetto al placebo.
** All Patients as Treated (APaT) Population, ad esclusione dei dati dopo un trattamento glicemico d'urgenza.
†† p=0,007 rispetto al placebo.
Sitagliptin come terapia add-on in pazienti non sufficientemente controllati con una combinazione di metformina non a rilascio prolungato e insulina
Complessivamente 641 pazienti affetti da diabete di tipo 2 hanno partecipato a uno studio di 24 settimane randomizzato, in doppio cieco e controllato con placebo, condotto per valutare l'efficacia di 100 mg di sitagliptin una volta al giorno in combinazione con una dose stabile di insulina. Il 75% circa dei pazienti assumeva contemporaneamente metformina. I pazienti trattati con insulina pronta all'uso a lunga durata d'azione o ad azione intermedia (con o senza metformina) sono stati randomizzati per ricevere in aggiunta 100 mg di sitagliptin o placebo. Gli endpoint glicemici misurati comprendevano HbA1c, glicemia a digiuno e glicemia postprandiale (PPG) a 2 ore.
La combinazione di sitagliptin, metformina e insulina ha indotto miglioramenti significativi dell'HbA1c, della glicemia a digiuno e della glicemia postprandiale (PPG) a 2 ore rispetto al placebo (Tabella 5). Il miglioramento dell'HbA1c rispetto al placebo è stato generalmente simile in tutti i sottogruppi definiti in base a sesso, età, etnia, indice di massa corporea (BMI) iniziale, tempo trascorso dalla diagnosi di diabete, presenza di sindrome metabolica, indizi standard di insulino-resistenza (HOMA-IR) o secrezione di insulina (HOMA-β). In nessuno dei gruppi è stata rilevata una variazione notevole del peso corporeo rispetto al valore iniziale.
Tabella 5
Parametri glicemici e peso corporeo rilevati nell'esame conclusivo (studio di 24 settimane) dopo l'assunzione di sitagliptin come terapia add-on in combinazione con metformina e una dose stabile di insulina†
Sitagliptin 100 mg | Placebo | |
HbA1c (%) | N = 223 | N = 229 |
Valore iniziale (medio) | 8,73 | 8,60 |
Variazione rispetto al basale (media aggiustata‡) | -0,66 | -0,13 |
Differenza rispetto al placebo (media aggiustata‡,§) | -0,53** | |
Pazienti (%) con HbA1c <7% | 32 (14,3) | 12 (5,2) |
Glicemia a digiuno (mmol/l) | N = 225 | N = 229 |
Valore iniziale (medio) | 9,5 | 9,7 |
Variazione rispetto al valore iniziale (media aggiustata‡) | -1,2 | -0,2 |
Differenza rispetto al placebo (media aggiustata‡) | -1,0** | |
PPG a 2 ore (mmol/l) | N = 182 | N = 189 |
Valore iniziale (medio) | 15,4 | 15,4 |
Variazione rispetto al valore iniziale (media aggiustata‡) | -2,1 | 0,1 |
Differenza rispetto al placebo (media aggiustata‡) | -2,2** | |
Peso corporeo (kg)¶ | N = 201 | N = 200 |
Valore iniziale (medio) | 87,9 | 88,0 |
Variazione rispetto al valore iniziale (media aggiustata‡) | -0,1 | 0,0 |
Differenza rispetto al placebo (media aggiustata‡) | 0,1# |
† All Patients Treated Population (analisi intention-to-treat).
‡ Medie dei minimi quadrati aggiustate per trattamento con insulina alla visita 1 (pronta all'uso verso non pronta all'uso [ad azione intermedia o a lunga durata d'azione]) e valore iniziale.
§ Stratificazione in funzione dell'insulina, l'interazione non è stata significativa (p>0,10).
** p<0,001 rispetto al placebo.
¶ All Patients as Treated (APaT) Population, ad esclusione dei dati dopo un trattamento glicemico d'urgenza.
# Statisticamente non significativo (p≥0,05) rispetto al placebo.
In un altro studio di 24 settimane randomizzato, in doppio cieco e controllato con placebo, condotto per valutare l'efficacia nel risparmio di insulina del sitagliptin come terapia add-on combinata, 660 pazienti con un controllo glicemico insufficiente con insulina glargine con o senza metformina (≥1500 mg al giorno) sono stati randomizzati a 100 mg di sitagliptin (N=330) o placebo (N=330). La somministrazione è avvenuta una volta al giorno durante un'intensificazione della terapia con insulina. Tra i pazienti che prendevano metformina, il valore basale della HbA1c era di 8,70% e la dose di insulina iniziale era di 37 UI/die. I pazienti venivano istruiti a titolare la dose di insulina glargine in base ai valori di glicemia a digiuno misurati mediante fingerstick. Gli endpoint secondari dello studio comprendevano HbA1c e FPG.
Tra i pazienti che prendevano metformina, alla settimana 24 l'aumento della dose giornaliera di insulina nei pazienti trattati con sitagliptin (19 UI/die, N=285) è stato inferiore rispetto ai pazienti trattati con placebo (24 UI/die, N=283). La differenza è risultata statisticamente significativa (p=0,007).
Riguardo agli endpoint secondari dello studio, la riduzione di HbA1c nei pazienti trattati con sitagliptin, metformina e insulina è stata di -1,35% rispetto a -0,90% nei pazienti trattati con placebo, metformina e insulina, pari a una differenza di -0,45% (IC al 95%: -0,62, -0,29). La riduzione dell'FPG nei pazienti trattati con sitagliptin, metformina e insulina è stata di ‑3,0 mmol/l rispetto a -2,4 mmol/l nei pazienti trattati con placebo, metformina e insulina, pari a una differenza di -0,7 mmol/l (IC al 95%: -1,0, -0,3).
Studio controllato con glipizide in combinazione con metformina non a rilascio prolungato
L'efficacia a lungo termine è stata esaminata in uno studio di 52 settimane randomizzato, in doppio cieco e controllato con glipizide, condotto su pazienti affetti da diabete di tipo 2. I pazienti con inadeguato controllo glicemico con la metformina (≥1500 mg/die) hanno ricevuto per 52 settimane 100 mg al giorno di sitagliptin (N=588) o di glipizide (N=584). Ai pazienti che hanno ricevuto glipizide è stata somministrata una dose iniziale di 5 mg/die, aumentata selettivamente in modo graduale nel corso delle 18 settimane seguenti, senza provocare un'ipoglicemia significativa, fino a raggiungere un valore target di FPG <110 mg/dl. Per un controllo glicemico ottimale era consentita una dose massima di 20 mg/die. Dopodiché la dose di glipizide è stata mantenuta costante. La dose media di glipizide dopo la titolazione è stata di 10,3 mg.
Entrambi i trattamenti hanno determinato un miglioramento statisticamente significativo del controllo glicemico rispetto al valore iniziale. Dopo 52 settimane, la riduzione del valore iniziale della HbA1c è stata di 0,67% per 100 mg di sitagliptin al giorno e di 0,67% per la glipizide, cosa che ha confermato l'equivalenza dei due principi attivi. La riduzione dell'FPG è stata di 0,56 mmol/l per il sitagliptin e 0,42 mmol/l per la glipizide. In un'analisi post-hoc, in entrambi i gruppi i pazienti con un valore iniziale della HbA1c più elevato (≥9%) hanno presentato una maggiore riduzione del valore iniziale della HbA1c (sitagliptin -1,68%; glipizide -1,76%). Il trattamento con sitagliptin ha determinato una riduzione del rapporto proinsulina/insulina un indice per la sintesi e il rilascio dell'insulina. Con la glipizide il rapporto proinsulina/insulina è peggiorato. L'incidenza dell'ipoglicemia era significativamente inferiore nel gruppo sitagliptin con 4,9% rispetto a quella nel gruppo glipizide (32,0%). I pazienti trattati con sitagliptin hanno mostrato una significativa diminuzione del peso corporeo rispetto a un significativo aumento di peso riscontrato nei pazienti in terapia con glipizide (-1,5 kg vs. +1,1 kg).
Metformina cloridrato
Lo studio prospettico randomizzato (UKPDS) ha confermato il beneficio a lungo termine di un controllo intensivo della glicemia nel diabete di tipo 2. L'analisi dei risultati effettuata in pazienti in sovrappeso trattati con metformina dopo che una dieta da sola non ha aiutato ha mostrato:
- nel gruppo di trattamento con metformina, una riduzione significativa del rischio assoluto di subire un complicanza correlata al diabete (29,8 eventi/1000 anni paziente) rispetto alla dieta da sola (43,3 eventi/1000 anni paziente), p=0,0023, e rispetto ai gruppi di trattamento combinati con sulfonilurea e insulina in monoterapia (40,1 eventi/1000 anni paziente), p=0,0034;
- una riduzione significativa del rischio assoluto di morte correlata al diabete: metformina 7,5 decessi/1000 anni paziente, dieta da sola 12,7 decessi/1000 anni paziente, p=0,017;
- una riduzione significativa del rischio assoluto di mortalità totale: metformina 13,5 decessi/1000 anni paziente, rispetto alla dieta da sola 20,6 decessi/1000 anni paziente (p=0,011) e rispetto ai gruppi di trattamento combinati con sulfonilurea e insulina in monoterapia 18,9 decessi/1000 anni paziente (p=0,021);
- una riduzione significativa del rischio assoluto di infarto cardiaco: metformina 11 eventi/1000 anni paziente, dieta da sola 18 decessi/1000 anni paziente (p=0,01).
Studio sulla sicurezza cardiovascolare TECOS
Lo studio TECOS (Trial Evaluating Cardiovascular Outcomes with Sitagliptin) era randomizzato e includeva 14'671 pazienti nella popolazione da trattare (intention-to-treat) con valori di HbA1c compresi tra ≥6,5 e 8,0% e con malattia cardiovascolare diagnosticata. I pazienti sono stati trattati con 100 mg al giorno di sitagliptin (7332) (o 50 mg al giorno se il valore iniziale di eGFR era ≥30 e <50 ml/min/1,73 m2) o placebo (7339), aggiunti alla terapia solitamente utilizzata per il raggiungimento dei valori standard regionali per l'HbA1c e per i fattori di rischio cardiovascolare. I pazienti con eGFR <30 ml/min/1,73 m2 non sono stati inclusi nello studio. Lo studio comprendeva 2004 pazienti di età ≥75 anni e 3324 pazienti con insufficienza renale (eGFR <60 ml/min/1,73 m2).
I pazienti del gruppo sitagliptin hanno ricevuto una quantità inferiore di medicamento anti-iperglicemico rispetto a quelli del gruppo placebo (Hazard Ratio 0,72; IC al 95%: 0,68-0,77; p≤0,001). Inoltre, nei pazienti che all'inizio dello studio non erano trattati con insulina, l'inizio di un'insulinoterapia cronica era meno probabile (Hazard Ratio 0,70; IC al 95%: 0,63-0,79; p<0,001).
L'endpoint primario cardiovascolare era un composito di prima insorgenza di morte cardiovascolare, infarto miocardico non fatale, ictus non fatale od ospedalizzazione per angina pectoris instabile. Gli endpoint secondari cardiovascolari comprendevano prima insorgenza di morte cardiovascolare, infarto miocardico non fatale o ictus non fatale; prima insorgenza dei singoli componenti dell'endpoint primario composito; mortalità totale e ospedalizzazione per insufficienza cardiaca.
Dopo un follow-up mediano di tre anni, il sitagliptin, quando aggiunto alla terapia solitamente utilizzata nei pazienti affetti da diabete di tipo 2, non ha aumentato il rischio di gravi eventi avversi cardiovascolari o il rischio di ospedalizzazione per insufficienza cardiaca (Tabella 6).
Tabella 6
Tassi degli eventi cardiovascolari e degli eventi secondari importanti
Sitagliptin 100 mg | Placebo | Hazard Ratio | Valore p† | |||
N (%) | Tasso d'incidenza per 100 anni paziente* | N (%) | Tasso d'incidenza per 100 anni paziente | |||
Analisi della popolazione da trattare (intention-to-treat) | ||||||
Numero di pazienti | 7332 | 7339 | 0,98 (0,89; 1,08) | <0,001 | ||
Endpoint primario composito (morte cardiovascolare, infarto miocardico non fatale, ictus non fatale od ospedalizzazione per angina pectoris instabile) | 839 (11,4) | 4,1 | 851 (11,6) | 4,2 | ||
Endpoint secondario composito (morte cardiovascolare, infarto miocardico non fatale o ictus non fatale) | 745 (10,2) | 3,6 | 746 (10,2) | 3,6 | 0,99 (0,89; 1,10) | <0,001 |
Eventi secondari | ||||||
Morte cardiovascolare | 380 (5,2) | 1,7 | 366 (5,0) | 1,7 | 1,03 (0,89; 1,19) | 0,711 |
Tutti gli infarti miocardici (fatali e non-fatali) | 300 (4,1) | 1,4 | 316 (4,3) | 1,5 | 0,95 (0,81; 1,11) | 0,487 |
Tutti gli ictus (fatali e non-fatali) | 178 (2,4) | 0,8 | 183 (2,5) | 0,9 | 0,97 (0,79; 1,19) | 0,760 |
Ospedalizzazione per angina pectoris instabile | 116 (1,6) | 0,5 | 129 (1,8) | 0,6 | 0,90 (0,70-1,16) | 0,419 |
Morte per qualsiasi causa | 547 (7,5) | 2,5 | 537 (7,3) | 2,5 | 1,01 (0,90; 1,14) | 0,875 |
Ospedalizzazione per insufficienza cardiaca‡ | 228 (3,1) | 1,1 | 229 (3,1) | 1,1 | 1,00 (0,83; 1,20) | 0,983 |
* Il tasso d'incidenza per 100 anni paziente è stato calcolato come 100 × (numero totale di pazienti con ≥1 evento durante il periodo di esposizione eleggibile per il totale degli anni paziente del follow-up).
† Basato su un modello di Cox stratificato per regione. Per gli endpoint compositi, i valori p si riferiscono a un test di non-inferiorità volto a dimostrare che il rapporto di rischio (Hazard Ratio) è inferiore a 1,3. Per tutti gli altri endpoint, i valori p si riferiscono a un test delle differenze tra tassi di rischio (Hazard Rates).
‡ L'analisi dell'ospedalizzazione per insufficienza cardiaca è stata aggiustata per l'anamnesi di insufficienza cardiaca all'entrata nello studio.
Farmacocinetica
I risultati di uno studio condotto su probandi sani ha dimostrato che le compresse di Janumet XR (sitagliptin fosfato/metformina cloridrato a rilascio prolungato) 50 mg/500 mg e 100 mg/1000 mg sono bioequivalenti alla somministrazione concomitante delle dosi corrispondenti di sitagliptin fosfato (Januvia) e metformina cloridrato a rilascio prolungato in compresse separate.
È stata dimostrata anche la bioequivalenza tra due compresse di Janumet XR 50 mg/500 mg e una compressa di Janumet XR 100 mg/1000 mg.
In uno studio crossover condotto su probandi sani, l'AUC e la Cmax del sitagliptin e l'AUC della metformina dopo la somministrazione di una sola compressa di Janumet XR 50 mg/500 mg (formulazione testata) sono risultate simili a quelle rilevate dopo la somministrazione di una sola compressa di Janumet (sitagliptin fosfato/metformina cloridrato non a rilascio prolungato) da 50 mg/500 mg. Dopo la somministrazione di una singola compressa di Janumet XR 50 mg/500 mg (formulazione testata), il valore medio della Cmax della metformina è stato inferiore del 30% e il valore mediano di Tmax 4 ore dopo rispetto ai valori corrispondenti osservati dopo la somministrazione di una singola compressa di Janumet 50 mg/500 mg. Ciò rispecchia le caratteristiche attese del rilascio modificato della metformina della formulazione di Janumet XR.
Dopo la somministrazione a probandi sani adulti di due compresse di Janumet XR 50 mg/1000 mg una volta al giorno per sette giorni insieme alla cena, sitagliptin e metformina hanno raggiunto lo steady state rispettivamente il giorno 4 e 5. I valori mediani di Tmax per sitagliptin e metformina allo steady state sono stati rispettivamente di ca. 3 e 8 ore dopo l'assunzione. I valori mediani di Tmax per sitagliptin e metformina dopo la somministrazione di una singola compressa di Janumet sono stati rispettivamente di ca. 3 e 3,5 ore dopo l'assunzione.
I dati farmacocinetici che caratterizzano la distribuzione, il metabolismo e l'eliminazione di metformina e sitagliptin provengono da studi eseguiti con la somministrazione dei singoli principi attivi metformina non a rilascio prolungato e sitagliptin e sono applicabili a Janumet XR.
Assorbimento
Janumet XR
Successivamente alla somministrazione di compresse Janumet XR con una colazione ad alto contenuto lipidico, la AUC del sitagliptin è risultata invariata. È stata riscontrata una riduzione del 17% della Cmax media, nonostante il valore mediano di Tmax fosse invariato rispetto allo stato a digiuno. Successivamente alla somministrazione di Janumet XR con una colazione ad alto contenuto lipidico, la AUC della metformina era aumentata del 62%, la Cmax della metformina era diminuita del 9% e il Tmax mediano della metformina era prolungato di 2 ore rispetto allo stato a digiuno.
Sitagliptin fosfato
La biodisponibilità assoluta del sitagliptin è pari a circa l'87%. La somministrazione concomitante di un pasto ad alto contenuto lipidico e sitagliptin fosfato non ha avuto effetti sulla farmacocinetica del sitagliptin.
Distribuzione
Sitagliptin fosfato
Allo steady state, il volume di distribuzione medio dopo una dose singola endovenosa di 100 mg di sitagliptin a probandi sani è di circa 198 litri. La frazione di sitagliptin legata in modo reversibile alle proteine plasmatiche è bassa (38%).
Metformina cloridrato
Non sono stati condotti studi sulla distribuzione della metformina a rilascio prolungato. Il volume di distribuzione apparente (V/F) della metformina dopo l'assunzione orale di dosi singole di 850 mg di metformina cloridrato non a rilascio prolungato era in media di 654 ± 358 l. Diversamente dalle sulfoniluree, che sono legate per più del 90% alle proteine plasmatiche, la metformina è scarsamente legata alle proteine plasmatiche. La metformina si ripartisce negli eritrociti, probabilmente in funzione del tempo. Alle dosi cliniche usuali e agli usuali schemi posologici per la metformina cloridrato in compresse, le concentrazioni plasmatiche della metformina allo steady state sono raggiunte entro 24-48 ore e sono generalmente <1 µg/ml. In studi clinici controllati con la metformina, i livelli plasmatici massimi della metformina non hanno mai superato i 5 µg/ml, anche ai dosaggi massimi.
Metabolismo
Sitagliptin fosfato
Il sitagliptin viene eliminato principalmente immodificato per via urinaria e solo una piccola frazione viene metabolizzata. Circa il 79% del sitagliptin viene escreto immodificato nelle urine.
Dopo la somministrazione di una dose di [14C]sitagliptin per via orale, circa il 16% della radioattività è stata escreta sotto forma di metaboliti del sitagliptin. Sono state rinvenute tracce di 6 metaboliti e non si presume che essi contribuiscano all'attività inibitoria del sitagliptin sulla DPP-4 plasmatica. Studi in vitro hanno indicato che l'enzima primariamente responsabile del limitato metabolismo del sitagliptin è il CYP3A4, con un contributo del CYP2C8.
Metformina cloridrato
Studi condotti con somministrazione di dosi singole endovenose a probandi sani hanno evidenziato che la metformina viene escreta immodificata nelle urine, non viene metabolizzata nel fegato (nell'uomo non sono stati identificati metaboliti) e non viene escreta per via biliare.
Eliminazione
Sitagliptin fosfato
Dopo la somministrazione di una dose di [14C]sitagliptin per via orale a probandi sani, circa il 100% della radioattività somministrata è stata eliminata nelle feci (13%) o nelle urine (87%) entro una settimana. L'emivita terminale apparente t1/2 dopo una dose di 100 mg di sitagliptin per via orale è stata di ca. 12,4 ore, mentre la clearance renale è stata di circa 350 ml/min.
L'eliminazione del sitagliptin ha luogo principalmente attraverso l'escrezione renale e implica una secrezione tubulare attiva. Il sitagliptin è un substrato del trasportatore anionico organico umano 3 (hOAT-3), che può essere implicato nell'eliminazione renale del sitagliptin. Non è stato ancora possibile dimostrare la rilevanza clinica di hOAT-3 nel trasporto del sitagliptin. Il sitagliptin è anche un substrato della glicoproteina-P, che potrebbe anche essere implicata nel processo di eliminazione renale del sitagliptin. Tuttavia la ciclosporina, un inibitore della glicoproteina-P, non ha ridotto la clearance renale del sitagliptin.
Metformina cloridrato
La clearance renale è ca. 3,5 volte maggiore di quella della creatinina, il che fa supporre che la secrezione tubulare rappresenti la più importante via di eliminazione della metformina. Dopo la somministrazione orale di metformina non a rilascio prolungato, il 90% circa del medicamento assorbito viene escreto per via renale entro le prime 24 ore, con un'emivita di eliminazione nel plasma di ca. 6,2 ore. Nel sangue, l'emivita di eliminazione è di ca. 17,6 ore, il che fa supporre che la massa eritrocitaria potrebbe essere un compartimento di distribuzione.
Disturbi della funzionalità renale
Sitagliptin fosfato
In pazienti con disturbo della funzionalità renale moderato, con un eGFR compreso tra 30 e <45 ml/min/1,73 m2, è stato osservato un aumento di ca. 2 volte dell'AUC plasmatica del sitagliptin, mentre in pazienti con un disturbo della funzionalità renale grave (eGFR <30 ml/min/1,73 m2), inclusi i pazienti con insufficienza renale terminale (end-stage renal disease, ESRD) in emodialisi, tale aumento è stato ca. 4 volte superiore rispetto a probandi con funzionalità renale normale.
Metformina cloridrato
Nei pazienti con funzionalità renale ridotta, l'emivita della metformina non a rilascio prolungato nel plasma e nel sangue è prolungata e la clearance renale è diminuita (vedere CONTOINDICAZIONI e AVVERTENZE E MISURE PRECAUZIONALI).
Disturbi della funzionalità epatica
Sitagliptin fosfato
Dopo la somministrazione di una dose singola di 100 mg di sitagliptin in pazienti con disturbo della funzionalità epatica moderato (punteggio di Child-Pugh compreso tra 7 e 9), l'AUC e la Cmax medie del sitagliptin sono aumentate rispettivamente di ca. il 21% e il 13% rispetto al gruppo di controllo di probandi sani. Queste differenze sono considerate clinicamente non rilevanti.
Non sono disponibili dati clinici per pazienti con disturbo della funzionalità epatica grave (punteggio di Child-Pugh >9). Poiché però il sitagliptin viene eliminato in primo luogo per via renale, i disturbi della funzionalità epatica gravi non dovrebbero compromettere la farmacocinetica del sitagliptin.
Metformina cloridrato
Nei pazienti con disturbo della funzionalità epatica grave non sono stati effettuati studi farmacocinetici con la metformina.
Uso negli anziani
Sitagliptin fosfato
In base a un'analisi di farmacocinetica di popolazione dei dati di fase I e di fase II, l'età non ha un impatto clinicamente significativo sulla farmacocinetica del sitagliptin. Negli anziani (da 65 a 80 anni) sono state osservate concentrazioni plasmatiche di sitagliptin superiori di circa il 19% rispetto ai giovani.
Metformina cloridrato
Sono disponibili solo pochi dati di studi farmacocinetici controllati sulla metformina non a rilascio prolungato condotti su soggetti anziani sani. Questi dati suggeriscono una ridotta clearance plasmatica complessiva della metformina, un'emivita prolungata e una Cmax aumentata negli anziani sani rispetto ai probandi sani giovani.
Questi dati fanno supporre che l'influenza dell'età del paziente sulla farmacocinetica della metformina sia da ricondurre essenzialmente all'alterazione della funzionalità renale (vedere l'informazione professionale di GLUCOPHAGE1).
1 GLUCOPHAGE® è un marchio registrato di Merck Sante S.A.S, società partner di Merck KGaA di Darmstadt, Germania.
Impiego nei bambini e negli adolescenti
La sicurezza e l'efficacia di Janumet XR nei bambini e negli adolescenti non sono state esaminate.
Sesso
Sitagliptin fosfato
In base a un'analisi di farmacocinetica dei dati di fase I e a un'analisi di farmacocinetica di popolazione dei dati di fase I e fase II, il sesso non ha un impatto clinicamente significativo sulla farmacocinetica del sitagliptin.
Metformina cloridrato
Se analizzati in funzione del sesso, i parametri farmacocinetici della metformina nei probandi sani e nei pazienti affetti da diabete di tipo 2 non differiscono sostanzialmente. Parimenti, in studi clinici controllati condotti su pazienti affetti da diabete di tipo 2 è stato possibile evidenziare che l'effetto anti-iperglicemico della metformina è risultato simile negli uomini e nelle donne.
Etnia
Sitagliptin fosfato
In base a un'analisi di farmacocinetica dei dati di fase I e a un'analisi di farmacocinetica di popolazione dei dati di fase I e fase II, con l'inclusione di bianchi, latino-americani, neri, asiatici e membri di altri gruppi etnici, l'etnia non ha un impatto clinicamente significativo sulla farmacocinetica del sitagliptin.
Metformina cloridrato
Non esistono studi sui parametri farmacocinetici della metformina in funzione dell'etnia. In studi clinici condotti con la metformina nei pazienti affetti da diabete di tipo 2, l'effetto anti-iperglicemico nei bianchi (n=249), nei neri (n=51) e nei latino-americani (n=24) è risultato comparabile.
Indice di massa corporea (Body-Mass-Index, BMI)
Sitagliptin fosfato
In base a un'analisi di farmacocinetica dei dati di fase I e a un'analisi di farmacocinetica di popolazione dei dati di fase I e fase II, l'indice di massa corporea non ha alcun impatto clinicamente significativo sulla farmacocinetica del sitagliptin.
Dati preclinici
Sitagliptin e metformina
Studi preclinici di tossicocinetica e studi di tossicità orale con sitagliptin e metformina come preparato combinato non a rilascio prolungato sono stati condotti sul cane.
In uno studio di tossicità orale di 16 settimane, a cani di sesso femminile sono stati somministrati 20 mg/kg/die di metformina da sola o in combinazione con 2, 10 o 50 mg/kg/die di sitagliptin. Nel gruppo ad alto dosaggio sono stati osservati atassia transitoria e/o tremore. Questi sintomi sono stati giudicati imputabili al sitagliptin, poiché sono stati osservati in altri studi su cani con sitagliptin da solo a un dosaggio di 50 mg/kg/die. In questo studio, il livello senza effetto (no-effect level) per le alterazioni imputabili al trattamento era di 10 mg/kg/die di sitagliptin e 20 mg/kg/die di metformina, pari a un'esposizione sistemica nell'uomo di circa 6 volte a una dose giornaliera di 100 mg di sitagliptin e a un'esposizione sistemica di 2,5 volte a una dose giornaliera di 2000 mg di metformina.
Con il preparato combinato Janumet XR non sono stati condotti studi preclinici riguardanti cancerogenesi, mutagenesi, compromissione della fertilità o effetti sulla riproduzione. I seguenti dati derivano da studi effettuati con sitagliptin o metformina separatamente.
Sitagliptin
In studi non clinici condotti su ratti sono stati osservati effetti del sitagliptin solo a esposizioni molto superiori alla dose usata nell'uomo, per cui non devono essere presi in considerazione per il normale impiego nell'uomo. In studi preclinici di sicurezza, il sitagliptin non si è rivelato né genotossico, né mutageno.
In una serie di studi di tossicità con somministrazione ripetuta del medicamento a cani, sono state esaminate dosi di 2, 10 e 50 mg/kg/die nel corso di un periodo fino a 53 settimane. Dopo 53 settimane di trattamento con una dose di 10 mg/kg/die, equivalente a circa 6 volte la dose di 100 mg/die raccomandata nell'essere umano adulto, non sono stati rilevati effetti. Nei cani trattati con 50 mg/kg/die di sitagliptin (equivalente a circa 26 volte il livello di esposizione nell'uomo), sono stati osservati sintomi fisici transitori legati alla terapia, quali ad esempio respirazione a bocca aperta, salivazione, emesi bianca schiumosa, atassia, tremore, diminuzione dell'attività e/o postura curva. Negli studi di tossicità di 14 e 27 settimane con una dose di 50 mg/kg/die è stata inoltre osservata a livello istologico una degenerazione lieve o molto lieve della muscolatura scheletrica. Nello studio di tossicità di 53 settimane non è stata tuttavia più rilevata alcuna degenerazione della muscolatura scheletrica, cosa che fa supporre che si sia trattato di un effetto non riproducibile o che questa alterazione non si sia più presentata nel corso di un trattamento più prolungato. L'esposizione sistemica nel range NOEL nello studio di 53 settimane (10 mg/kg/die) era di 5 volte superiore rispetto a quella nello studio di 27 settimane (2 mg/kg/die).
Ulteriori studi di tossicità per somministrazione orale di tre mesi sono stati condotti nelle scimmie rhesus e cynomolgus. Lo studio di 3 mesi condotto sulle scimmie rhesus ha esaminato il potenziale del sitagliptin di causare lesioni cutanee o tossicità renale; la valutazione si è limitata alla pelle e ai reni. Gli animali hanno ricevuto fino a 100 mg di sitagliptin/kg/die (pari a 28 volte il livello di esposizione sistemica con una dose quotidiana di 100 mg). Non ci sono stati rilievi ante mortem e post mortem imputabili al trattamento.
Nello studio di tossicità orale di 3 mesi condotto sulle scimmie cynomolgus sono state eseguite le valutazioni di routine complete. Gli animali hanno ricevuto fino a 100 mg di sitagliptin/kg/die (pari a 27 volte il livello di esposizione sistemica con una dose quotidiana di 100 mg). Non ci sono stati rilievi ante mortem e post mortem imputabili al trattamento.
Il sitagliptin non è risultato cancerogeno nei topi ai quali era stata somministrata per 2 anni per via orale la massima dose tollerabile di 500 mg/kg/die. Uno studio di cancerogenesi di due anni è stato condotto su ratti di sesso maschile e femminile ai quali sono state somministrate dosi orali di 50, 150 e 500 mg/kg/die di sitagliptin. Si è osservato un aumento dell'incidenza di adenomi e carcinomi epatici nei ratti di sesso maschile trattati con dose elevata e di carcinomi epatici in quelli di sesso femminile trattati con dose elevata. In base alla dose giornaliera raccomandata nell'uomo di 100 mg/die, questa dose nei ratti corrispondeva a ca. 58 volte l'esposizione nell'uomo. Negli esperimenti sui ratti questa dose era associata a epatotossicità. Il No-observed-Effect-Level per l'induzione di una neoplasia epatica è stato di 150 mg/kg/die, che corrisponde a ca. 19 volte l'esposizione nell'uomo alla dose raccomandata di 100 mg. Poiché è stato dimostrato che l'epatotossicità è correlata all'induzione di neoplasie epatiche nel ratto, questa più elevata incidenza di tumori epatici nel ratto è probabilmente la conseguenza della tossicità epatica cronica che si verifica a queste dosi elevate. La significatività clinica di questo risultato per l'uomo non è conosciuta.
In ratti maschi e femmine trattati prima e durante l'accoppiamento con dosi orali di sitagliptin fino a 1000 mg/kg/die (pari a ca. 100 volte l'esposizione nell'uomo, in base alla dose giornaliera raccomandata nell'essere umano adulto di 100 mg/die), non sono stati osservati effetti avversi sulla fertilità.
Studi di tossicità riproduttiva su ratti trattati con dosi orali di 1000 mg/kg/die hanno rilevato nella prole un lieve aumento dell'incidenza di malformazioni fetali delle costole (costole assenti, ipoplasiche e ondulate) correlato al trattamento. Il No-observed-Effect-Level per i disturbi dello sviluppo è stato di 250 mg/kg/die (pari a 32 volte l'esposizione nell'uomo, in base alla dose giornaliera raccomandata nell'essere umano adulto di 100 mg/die). Il sitagliptin viene escreto nel latte di ratti in allattamento.
Non sono stati condotti studi con sitagliptin, metformina e sulfonilurea.
Metformina cloridrato
I dati preclinici di studi convenzionali di farmacologia di sicurezza, tossicità a dosi ripetute del medicamento, genotossicità, potenziale carcinogeno e tossicità riproduttiva non mostrano rischi particolari per l'uomo correlati all'assunzione di metformina.
Altre indicazioni
Stabilità
Il medicamento non deve essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Precauzioni particolari per la conservazione
Tenere fuori dalla portata dei bambini.
Non conservare a temperature superiori a 30 °C. Tenere ben chiuso il flacone, per proteggere il contenuto dall'umidità.
Numero dell'omologazione
65052 (Swissmedic).
Titolare dell’omologazione
MSD MERCK SHARP & DOHME AG, Lucerna.
Stato dell'informazione
Settembre 2020.
HMV4+rhabdomyolysis/RCN000010607-CH+RCN000019721-CH
Composition
Principes actifs
Phosphate monohydraté de sitagliptine, chlorhydrate de metformine
Excipients
Povidone K29-32, hypromellose, hydroxypropylcellulose, silice colloïdale anhydre, fumarate de stéaryle sodique (chaque comprimé de 50 mg/500 mg contient 1,26 mg de sodium; chaque comprimé de 50 mg/1000 mg contient 1,78 mg de sodium; chaque comprimé de 100 mg/1000 mg contient 1,77 mg de sodium), gallate de propyle (E310), macrogol 3350, kaolin, dioxyde de titane (E171) et cire de carnauba et indigotine (E132).
Les comprimés Janumet XR 50 mg/500 mg contiennent également de la cellulose microcristalline. Les comprimés Janumet XR 50 mg/1000 mg contiennent également de l'oxyde de fer jaune (E172).
Forme pharmaceutique et quantité de principe actif par unité
Comprimé à libération modifiée contenant 50 mg de sitagliptine sous forme de phosphate monohydraté de sitagliptine et 500 mg de chlorhydrate de metformine à libération retardée (Janumet XR 50 mg/500 mg) ou 1000 mg de chlorhydrate de metformine à libération retardée (Janumet XR 50 mg/1000 mg), ainsi que contenant 100 mg de sitagliptine sous forme de phosphate monohydraté de sitagliptine et 1000 mg de chlorhydrate de metformine à libération retardée (Janumet XR 100 mg/1000 mg).
Indications/Possibilités d’emploi
Janumet XR est indiqué en tant que traitement complémentaire, en plus d'un régime alimentaire et d'une activité physique régulière, pour équilibrer la glycémie chez les patients diabétiques de type 2 lorsqu'un contrôle suffisant de la glycémie n'est atteint ni avec la metformine seule, ni avec la sitagliptine seule, ainsi que chez les patients déjà traités par une association de sitagliptine et de metformine.
Janumet XR est également indiqué en association avec une sulfonylurée (triple association thérapeutique), en plus d'un régime alimentaire et d'une activité physique régulière, chez les patients atteints de diabète de type 2 chez lesquels un contrôle suffisant de la glycémie n'est pas atteint par un traitement associant deux des trois principes actifs suivants: metformine, sitagliptine ou sulfonylurée.
Lorsqu'un régime alimentaire, une augmentation de l'activité physique et l'insuline ne permettent pas à eux seuls de diminuer suffisamment la glycémie: en association avec une insulinothérapie.
Posologie/Mode d’emploi
Généralités
La dose du traitement anti-hyperglycémiant par Janumet XR doit être définie de façon individualisée en fonction du traitement actuel du patient, de l'efficacité et de la tolérance, en veillant à ne pas dépasser la dose maximale recommandée de 100 mg de sitagliptine par jour.
Janumet XR doit être pris une fois par jour avec un repas, de préférence le soir.
La dose doit être augmentée progressivement afin de réduire le risque de troubles gastro-intestinaux dus à la metformine.
Pour que les propriétés de libération retardée restent préservées, les comprimés doivent être avalés entiers sans être divisés, cassés, broyés ou mâchés.
Les concentrations plasmatiques de metformine augmentent lors d'une prise de Janumet XR avec des aliments. Des cas de comprimés de Janumet XR non entièrement dissouts, excrétés dans les selles, ont été rapportés. On ne sait pas si la matière retrouvée dans les selles contient de la substance active. L'équilibre glycémique doit donc être contrôlé de manière particulièrement attentive chez les patients observant de manière répétée des restes de comprimés dans leurs selles.
Des modifications de traitements chez le diabète de type 2 exigent toujours de la précaution et une surveillance adéquate en raison des modifications possibles du contrôle glycémique. Ceci s'applique aussi au passage de comprimés de metformine à libération non retardée à des comprimés de metformine à libération retardée tels que p.ex. Janumet XR.
Recommandations posologiques
La dose initiale de Janumet XR doit se baser sur le traitement déjà suivi.
Janumet XR est pris une fois par jour avec un repas, de préférence le soir. Les comprimés Janumet XR sont disponibles dans les dosages suivants:
50 mg de sitagliptine/500 mg de chlorhydrate de metformine à libération retardée
50 mg de sitagliptine/1000 mg de chlorhydrate de metformine à libération retardée
100 mg de sitagliptine/1000 mg de chlorhydrate de metformine à libération retardée
Les patients utilisant les comprimés de 50 mg de sitagliptine/500 mg de metformine à libération retardée ou les comprimés de 50 mg de sitagliptine/1000 mg de metformine à libération retardée doivent prendre leur dose quotidienne en une seule prise de deux comprimés à la fois. Les comprimés de 100 mg de sitagliptine/1000 mg de metformine à libération retardée doivent être pris en un seul comprimé une fois par jour.
Patients chez lesquels un traitement à la metformine seule ne permet pas d'atteindre un équilibre glycémique suffisant
Chez les patients n'ayant pas atteint un équilibre glycémique suffisant sous metformine seule, la dose quotidienne initiale recommandée de Janumet XR est de 100 mg de sitagliptine au total plus la dose de metformine prescrite dans le cadre de l'ancien traitement.
Patients chez lesquels un traitement à la sitagliptine seule ne permet pas d'atteindre un équilibre glycémique suffisant
Chez les patients n'ayant pas atteint un équilibre glycémique suffisant sous sitagliptine seule, la dose quotidienne initiale recommandée de Janumet XR est de 100 mg de sitagliptine au total et de 1000 mg de chlorhydrate de metformine. La dose de metformine peut être ajustée selon le besoin conformément au contrôle glycémique atteint. Une augmentation progressive de la dose devrait être envisagée pour réduire les effets indésirables gastro-intestinaux éventuellement causés par la metformine. Les patients sous sitagliptine seule dont la dose a été ajustée en raison d'un trouble de la fonction rénale ne doivent pas passer à Janumet XR (voir la section «Contre-indications»).
Patients passant à Janumet XR après un traitement associé de sitagliptine et metformine
Les patients passant à Janumet XR après avoir été traités avec une association de sitagliptine et de metformine peuvent continuer à prendre la même dose initiale de sitagliptine et de metformine qu'auparavant sous la forme de l'association fixe Janumet XR.
Patients chez lesquels un traitement associant deux des trois antidiabétiques metformine, sitagliptine ou sulfonylurée ne permet pas d'atteindre un équilibre glycémique suffisant
La dose initiale usuelle de Janumet XR doit contenir au total 100 mg de sitagliptine par jour. Pour la définition de la dose initiale de la composante de metformine, il faut tenir compte de la glycémie actuelle et de la dose de metformine utilisée jusque-là le cas échéant. La dose de metformine doit être augmentée de façon progressive pour éviter des symptômes gastro-intestinaux dus à la metformine. Chez les patients qui prennent déjà ou doivent commencer à prendre une sulfonylurée, il est recommandé d'utiliser une dose réduite de la sulfonylurée afin de réduire le risque d'hypoglycémie induite par la sulfonylurée (voir «Mises en garde et précautions»).
Patients chez lesquels un traitement associant deux des trois antidiabétiques sitagliptine, metformine ou insuline ne permet pas d'atteindre un équilibre glycémique suffisant
La dose initiale usuelle de Janumet XR doit contenir au total 100 mg de sitagliptine par jour. Pour la définition de la dose initiale de la composante de metformine, il faut tenir compte de la glycémie actuelle et de la dose de metformine utilisée jusque-là le cas échéant. La dose de metformine doit être augmentée de façon progressive pour éviter d'éventuels symptômes gastro-intestinaux dus à la metformine. Chez les patients qui utilisent déjà ou doivent commencer à utiliser de l'insuline, il est recommandé d'utiliser une dose réduite d'insuline afin de diminuer le risque d'hypoglycémie (voir «Mises en garde et précautions»).
Aucune étude spécifique n'a été effectuée sur la sécurité et l'efficacité de Janumet XR chez les patients traités antérieurement par d'autres antidiabétiques oraux, qui sont ensuite passés à Janumet XR. Tout changement du traitement du diabète de type 2 doit être réalisé avec prudence et être soigneusement contrôlé, étant donné que des fluctuations de l'équilibre glycémique peuvent se produire.
Utilisation chez des patients souffrant d'un trouble de la fonction rénale
On évaluera la fonction rénale avant le début du traitement avec Janumet XR et ensuite à intervalles réguliers.
Janumet XR est contre-indiqué chez les patients dont le débit de filtration glomérulaire estimé (DFGe) est <30 ml/min/1,73 m2. Janumet XR doit être arrêté si le DFGe du patient s'abaisse plus tard en-dessous de 30 ml/min/1,73 m2 (voir «Contre-indications» et «Mises en garde et précautions»).
Chez des patients avec un DFGe ≥30 ml/min/1,73 m2 et <45 ml/min/1,73 m2 le commencement d'un traitement par Janumet XR n'est pas recommandé. Chez des patients qui prennent Janumet XR et chez lesquels le DFGe s'abaisse plus tard en-dessous de 45 ml/min/1,73 m2, le bénéfice et le risque de la continuation du traitement sont à évaluer et la dose de la sitagliptine est à limiter à 50 mg une fois par jour.
Utilisation chez les enfants et les adolescents
La sécurité et l'efficacité de Janumet XR n'ont pas été étudiées chez les enfants et les adolescents.
Utilisation chez les patients âgés
La sitagliptine et la metformine étant essentiellement éliminées par voie rénale, Janumet XR doit être utilisé avec d'autant plus de prudence que le patient est âgé. Un contrôle de la fonction rénale est en particulier nécessaire chez les patients âgés afin de prévenir une acidose lactique due à la metformine (voir la section «Mises en garde et précautions», Chlorhydrate de metformine, Acidose lactique).
Contre-indications
Janumet XR est contre-indiqué chez les patients présentant les particularités suivantes:
- Trouble sévère de la fonction rénale (DFGe <30 ml/min/1,73 m2) (voir «Mises en garde et précautions», Chlorhydrate de metformine, Trouble de la fonction rénale).
- Maladies aiguës susceptibles d'entraîner une détérioration de la fonction rénale, par exemple problèmes cardio-vasculaires (y compris infarctus du myocarde), septicémie, déshydratation (diarrhée, vomissements répétés), infections graves, par ex. des voies urinaires, forte fièvre.
- Hypersensibilité connue au phosphate de sitagliptine, au chlorhydrate de metformine ou à un quelconque composant de Janumet XR. (Voir les sections «Mises en garde et précautions», Réactions d'hypersensibilité, et «Effets indésirables», Expériences post-commercialisation).
- Acidose métabolique aiguë ou chronique, y compris acidocétose diabétique avec ou sans coma.
- L'application intravasculaire de produits de contraste contenant de l'iode pour des examens radiologiques peut entraîner une défaillance rénale et, de ce fait, causer une accumulation de metformine et une acidose lactique. Le traitement par la metformine doit être suspendu 48 h avant un tel examen si la clairance de la créatinine est <60 ml/min ou le DFGe <60 ml/min/1,73 m2. Le traitement avec la metformine ne pourra être poursuivi que si un contrôle de la fonction rénale réalisé 48 h après l'examen avec le produit de contraste ne montre pas de détérioration supplémentaire (voir «Mises en garde et précautions»).
Mises en garde et précautions
Janumet XR
Janumet XR ne devrait pas être utilisé chez les patients atteints de diabète de type 1 ou pour le traitement de l'acidocétose diabétique.
Pancréatite: Des cas de pancréatite aiguë, y compris des cas de pancréatite hémorragique ou nécrosante avec issue fatale et non fatale (voir «Effets indésirables»), ont été observés chez des patients sous sitagliptine. Les patients doivent être informés sur les symptômes caractéristiques d'une pancréatite aiguë: douleurs abdominales sévères, persistantes. Une amélioration de la pancréatite a été observée après l'arrêt du traitement par la sitagliptine. En cas de suspicion de pancréatite, on arrêtera le traitement par Janumet XR et par d'autres médicaments potentiellement suspects.
Contrôle de la fonction rénale: La sitagliptine et la metformine sont essentiellement éliminées par voie rénale. Avant le début d'un traitement avec Janumet XR, la fonction rénale doit être examinée car le risque d'accumulation de la metformine et d'acidose lactique augmente avec la limitation de la fonction rénale. Janumet XR est contre-indiqué chez les patients qui présentent un trouble grave de la fonction rénale (DFGe <30 ml/min/1,73 m2).
La fonction rénale doit être déterminée à intervalles réguliers à nouveau sous traitement par Janumet XR, soit au minimum:
- une fois par an chez les patients dont la fonction rénale est normale;
- au moins tous les 3 à 6 mois chez les patients dont le DFGe est ≥45 - 59 ml/min/1,73 m2, ainsi que chez les patients âgés;
- au moins tous les 3 mois chez les patients dont le DFGe est ≥30 - 44 ml/min/1,73 m2.
La prise de Janumet XR soit être arrêtée immédiatement si le DFGe diminue en-dessous de 30 ml/min/1,73 m2.
En association avec des agents susceptibles de provoquer une hypoglycémie: On sait que des antidiabétiques de la classe des sulfonylurées ou de l'insuline peuvent provoquer des hypoglycémies. Pour cette raison, il peut être nécessaire de réduire la posologie de l'insuline ou de la sulfonylurée en cas d'association avec Janumet XR.
Phosphate de sitagliptine
Myopathie/rhabdomyolyse
En rapport avec l'utilisation de Janumet XR des myopathies ont été signalées qui se manifestent par des myalgies, une faiblesse musculaire ou une sensibilité musculaire, accompagnées d'un taux de créatine kinase fortement augmenté (CK, jusqu'à dix fois la limite supérieure de la norme). La myopathie peut parfois se manifester sous la forme d'une rhabdomyolyse, accompagnée ou non d'une défaillance rénale aiguë suite à une myoglobinurie et des décès ont eu lieu dans de rares cas.
La prescription de Janumet XR à des patients présentant des facteurs prédisposant à une rhabdomyolyse requiert une prudence particulière. Une détermination du taux de créatine kinase devrait être réalisée avant le début du traitement dans les situations suivantes:
- Insuffisance rénale.
- Hypothyroïdie non contrôlée.
- Antécédents personnels ou familiaux de maladies musculaires héréditaires.
- Antécédents de toxicité musculaire avec une statine ou un fibrate.
- Dépendance à l'alcool.
- Chez les personnes âgées (≥65 ans), la nécessité d'une telle mesure devrait être envisagée en présence d'autres facteurs prédisposant à une rhabdomyolyse.
- Chez les personnes de sexe féminin, la nécessité d'une telle mesure devrait être envisagée en présence d'autres facteurs prédisposant à une rhabdomyolyse.
Dans ces situations, le risque d'un traitement devrait être considéré dans l'optique du bénéfice escompté.
En association avec des agents susceptibles de provoquer une hypoglycémie: Dans les études cliniques sur l'administration de sitagliptine seule ou en association avec des principes actifs non associés à une hypoglycémie (par exemple metformine ou agonistes du PPARγ (thiazolidinediones)), l'incidence des hypoglycémies en rapport avec la prise de sitagliptine a été similaire à celle observée sous placebo.
Réactions d'hypersensibilité
Il existe des rapports post-commercialisation de réactions sérieuses d'hypersensibilité chez des patients traités à la sitagliptine (l'un des principes actifs de Janumet XR). Ces réactions ont englobé: anaphylaxie, angio-œdème et anomalies cutanées exfoliatives, y compris syndrome de Stevens-Johnson. Ces réactions se sont produites dans les 3 premiers mois du traitement à la sitagliptine, parfois même après la première dose. Étant donné que ces rapports proviennent d'une population de taille inconnue, il est impossible de fournir des indications fiables sur la fréquence de ces événements. Lors d'une suspicion de réaction d'hypersensibilité, le traitement par Janumet XR doit être arrêté (Voir la section «Contre-indications» et «Effets indésirables», Expériences post-commercialisation).
Pemphigoïde bulleuse
Après commercialisation, des cas de pemphigoïde bulleuse ayant nécessité une hospitalisation ont été rapportés avec l'utilisation d'inhibiteurs de la dipeptidylpeptidase-4 (DPP-4). Dans les cas rapportés, les patients ont récupéré généralement sous traitement immunosuppresseur topique ou systémique et après arrêt de la prise de l'inhibiteur de la DPP-4. Il faut signaler aux patients de contacter leur médecin en cas d'apparition de vésicules ou d'érosions cutanées sous traitement par Janumet XR. En cas de suspicion de pemphigoïde bulleuse, le traitement par Janumet XR devrait être arrêté. Il faudrait considérer d'adresser le patient à un dermatologue pour confirmer le diagnostic et instaurer un traitement adéquat.
Chlorhydrate de metformine
Acidose lactique: L'acidose lactique est une complication métabolique rare mais grave pouvant être provoquée par une accumulation de metformine pendant le traitement par Janumet XR (phosphate de sitagliptine/chlorhydrate de metformine à libération retardée). L'acidose lactique est mortelle dans environ 50% des cas. L'acidose lactique peut aussi se produire sous différentes conditions physiopathologiques, y compris le diabète sucré, et chaque fois qu'une hypoperfusion et une hypoxie tissulaire se développent dans une mesure significative. L'acidose lactique est caractérisée par un taux sanguin accru de lactate (>5 mmol/l), un pH sanguin abaissé, des troubles électrolytiques avec trou anionique augmenté et une valeur accrue du rapport lactate/pyruvate. Lorsque la metformine est à l'origine d'une acidose lactique, on trouve généralement des taux plasmatiques de metformine supérieurs à 5 µg/ml.
L'incidence de l'acidose lactique chez les patients traités à la metformine est très faible (environ 0,03 cas sur 1000 patient-années, avec environ 0,015 cas de décès sur 1000 patient-années). Dans des études cliniques sur plus de 20'000 patient-années d'exposition à la metformine, aucune acidose lactique n'a été rapportée. Les cas signalés se sont produits essentiellement chez des patients diabétiques avec une grave limitation de la fonction rénale ou une défaillance rénale chronique. Les patients insuffisants cardiaques qui nécessitent un traitement médicamenteux – en particulier les patients présentant une insuffisance cardiaque instable ou aiguë avec hypoperfusion et hypoxie – ont un risque accru de développer une acidose lactique. Le risque d'acidose lactique augmente avec le degré de l'insuffisance rénale et avec l'âge du patient. Ce risque peut donc significativement être abaissé par des contrôles réguliers de la fonction rénale chez les patients sous metformine, ainsi qu'en utilisant la plus faible dose efficace de metformine. La fonction rénale doit être surveillée soigneusement en particulier pendant le traitement des patients âgés (voir Utilisation chez des personnes âgées, Chlorhydrate de metformine). La metformine ne doit pas non plus être utilisée en présence de toute condition médicale associée à une hypoxie, une déshydratation ou une septicémie. Une fonction hépatique insuffisante pouvant significativement ralentir la dégradation du lactate, un traitement à la metformine doit en général être évité chez tous les patients chez lesquels le tableau clinique ou les résultats de laboratoire font suspecter une maladie hépatique.
Avant de prendre de la metformine, les patients doivent être mis en garde contre une consommation excessive aiguë ou chronique d'alcool, étant donné que l'alcool renforce les effets du chlorhydrate de metformine sur le métabolisme du lactate. En outre, le traitement à la metformine doit être suspendu transitoirement avant l'administration intravasculaire de substances de contraste et avant une intervention chirurgicale.
Le début de l'acidose lactique est souvent insidieux, avec des symptômes d'abord peu spécifiques tels que malaise, myalgies, détresse respiratoire, somnolence accrue et douleurs abdominales non spécifiques. Lors d'une acidose prononcée, on observera éventuellement une hypothermie, une hypotension et une brady-arythmie résistante. Le patient et le médecin traitant doivent être conscients de la signification éventuelle de tels symptômes et le patient doit être instruit de contacter le médecin immédiatement si de tels symptômes se manifestent. Dans un tel cas, le traitement à la metformine doit être arrêté et le patient doit être hospitalisé. Les électrolytes et cétones sériques, la glycémie et le pH sanguin peuvent être utiles pour le diagnostic, de même que les taux de lactate et de metformine.
Chez les patients sous metformine, des taux plasmatiques à jeun de lactate situés au-dessus de la normale mais au-dessous de 5 mmol/l n'évoquent pas nécessairement une acidose lactique menaçante et peuvent être expliqués par d'autres mécanismes tels que, par exemple, un diabète mal contrôlé ou un excès de poids, ou encore par un effort physique intensif ou des problèmes techniques lors de la manipulation des échantillons.
La possibilité d'une acidose lactique doit être considérée chez tout patient diabétique présentant une acidose métabolique en l'absence d'indices d'une acidocétose (cétonurie et cétonémie).
L'acidose lactique est une urgence médicale qui doit être traitée à l'hôpital. Chez un patient sous metformine qui présente une acidose lactique, le médicament doit être arrêté immédiatement et les mesures de soutien usuelles doivent être prises. Étant donné que le chlorhydrate de metformine et le lactate sont dialysables (avec une clairance allant jusqu'à 170 ml/min dans de bonnes conditions hémodynamiques), une hémodialyse immédiate est recommandée. Un tel traitement permet souvent une régression rapide des symptômes et un net rétablissement du patient (voir la section «Contre-indications»).
Une fois qu'un patient est stabilisé sous metformine, les symptômes gastro-intestinaux dus à la metformine – fréquents au début du traitement – sont très rares. Si des symptômes gastro-intestinaux apparaissent encore ultérieurement, ils peuvent être dus à une acidose lactique ou à une autre maladie sérieuse.
Hypoglycémie: Chez les patients traités à la metformine seule, aucune hypoglycémie n'est observée dans le cadre d'une utilisation normale. Dans des cas exceptionnels, une hypoglycémie peut se développer en association avec la metformine, par ex. en cas d'apport calorique insuffisant ou de dépense énergétique fortement accrue (grand effort physique), ainsi qu'en cas de consommation concomitante d'alcool ou d'autres substances hypoglycémiantes (par exemple sulfonylurées et insuline).
Les patients âgés, faibles ou présentant une malnutrition ou souffrant d'une insuffisance surrénalienne ou hypophysaire ou d'une intoxication alcoolique sont particulièrement prédisposés à subir des effets hypoglycémiques. L'hypoglycémie est parfois difficile à reconnaître chez les personnes âgées et chez les personnes prenant des antagonistes des récepteurs bêta-adrénergiques.
Prise concomitante d'autres médicaments pouvant influencer la fonction rénale ou la disponibilité de la metformine: Les médications concomitantes susceptibles d'influencer la fonction rénale ou de modifier significativement l'hémodynamique ou la disponibilité de la metformine – par exemple médicaments cationiques éliminés par sécrétion tubulaire dans le rein (voir la section «Interactions», Chlorhydrate de metformine) – doivent être utilisées avec prudence.
Utilisation de produits de contraste iodés: Les produits de contraste iodés administrés par voie intravasculaire pour des examens radiologiques peuvent entraîner une défaillance rénale. Comme cela peut mener à une accumulation de metformine et à une acidose lactique, le traitement par la metformine chez des patients dont le DFGe est <60 ml/min/1.73 m2 doit donc être suspendu à temps (environ 2 jours) et ne doit être repris que si la fonction rénale ne s'est pas détériorée 2 jours après l'examen avec le produit de contraste.
Hypoxie: Des défaillances cardio-vasculaires d'origines variées, insuffisances cardiaques aiguës, infarctus myocardiques aigus et autres états associés à une hypoxie ont été mis en rapport avec le développement d'une acidose lactique. Ces états peuvent aussi provoquer une azotémie prérénale. Si de tels événements se produisent pendant le traitement par Janumet XR, la prise du médicament doit immédiatement être arrêtée.
Opérations: La prise de Janumet XR doit transitoirement être suspendue pour les interventions chirurgicales (à l'exception d'interventions mineures permettant un apport de nourriture et de liquides sans restrictions) et n'être reprise qu'une fois qu'une absorption normale de nourriture est à nouveau possible et que la fonction rénale a été jugée suffisante (voir «Posologie/Mode d'emploi»).
Consommation d'alcool: On sait que l'alcool renforce les effets de la metformine sur le métabolisme du lactate. Les patients qui prennent Janumet XR doivent donc être mis en garde contre une consommation excessive – chronique ou aiguë – d'alcool.
Sodium: Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d. qu'il est essentiellement «sans sodium».
Insuffisance hépatique: L'insuffisance hépatique ayant été mise en rapport avec certains cas d'acidose lactique, Janumet XR ne doit pas être utilisé chez les patients chez lesquels le tableau clinique ou les résultats de laboratoire font soupçonner une maladie hépatique.
Taux sanguin de vitamine B12: Dans des études cliniques contrôlées sur l'administration de metformine pendant 29 semaines, environ 7% des patients ont présenté une réduction du taux de vitamine B12 à des valeurs inférieures à la moyenne alors que les valeurs initiales avaient été normales. Toutefois, cette réduction n'a pas été associée à des manifestations cliniques. Une telle réduction du taux sanguin de vitamine B12, probablement due à une réduction de l'absorption de la vitamine B12, n'est cependant que très rarement associée à une anémie et semble être rapidement réversible dès que l'administration de metformine est arrêtée ou que des suppléments de vitamine B12 sont administrés. Un examen annuel des paramètres hématologiques est recommandé chez tous les patients sous Janumet XR et tous les écarts doivent être soigneusement examinés et traités.
Les personnes dont l'apport ou l'absorption de vitamine B12 ou de calcium sont inadéquats semblent prédisposées à développer des taux sanguins réduits de vitamine B12. Chez ces patients, une vérification systématique du taux sérique de vitamine B12 tous les 2 à 3 ans est indiquée.
Modifications de l'état clinique des patients dont le diabète de type 2 était antérieurement sous contrôle: Un patient dont le diabète de type 2 était antérieurement bien contrôlé sous Janumet XR mais qui présente soudainement des valeurs de laboratoire anormales ou devient cliniquement malade (souvent de façon vague et mal définie) doit immédiatement être examiné pour identifier ou exclure une éventuelle acidocétose ou acidose lactique.
L'examen doit englober les électrolytes sériques, les cétones sériques et la glycémie ainsi que, si indiqué, le pH sanguin et les taux de lactate, de pyruvate et de metformine. En cas de suspicion d'acidose métabolique – quelle qu'en soit la genèse – la prise de Janumet XR doit être immédiatement arrêtée et le patient doit être hospitalisé.
Perte de contrôle de la glycémie: Chez les patients qui maintiennent un bon équilibre glycémique sous un quelconque antidiabétique, il est possible d'observer un dérèglement transitoire de l'équilibre glycémique dans des situations de stress telles que fièvre, traumatisme, infection ou opération.
Dans de tels cas, il sera éventuellement nécessaire de suspendre la prise de Janumet XR et d'administrer transitoirement de l'insuline à la place. Le traitement par Janumet XR pourra ensuite être poursuivi après la fin de l'épisode aigu.
Utilisation chez l'enfant et l'adolescent
La sécurité et l'efficacité de Janumet XR et de l'un de ses principes actifs, la sitagliptine, n'ont pas été étudiées chez les enfants et les adolescents de moins de 18 ans.
Utilisation chez les personnes âgées
Janumet XR
La sitagliptine et la metformine sont essentiellement éliminées par voie rénale. Janumet XR doit par conséquent être utilisé avec d'autant plus de prudence que le patient est âgé. Un contrôle de la fonction rénale est en particulier nécessaire chez les patients âgés afin de prévenir une acidose lactique due à la metformine (voir la section «Mises en garde et précautions», Chlorhydrate de metformine, Acidose lactique).
Phosphate de sitagliptine
Dans les études cliniques, la sécurité et l'efficacité de la sitagliptine ont été similaires chez les patients âgés (≥65 ans) et les patients plus jeunes (<65 ans).
Chlorhydrate de metformine
Les études cliniques contrôlées sur la metformine n'ont pas inclus suffisamment de patients âgés pour permettre de trancher définitivement la question de savoir si ces patients réagissent autrement que les patients plus jeunes. Toutefois, d'autres expériences cliniques n'ont révélé aucune différence entre patients âgés et patients plus jeunes.
Janumet XR doit en principe être pris avec le repas du soir. Une prise avec le repas permet de réduire en partie les effets indésirables gastro-intestinaux de la metformine. Il faut aussi compter que la biodisponibilité de la metformine à libération retardée est réduite dans le cas d'une prise à jeun, ce qui pourrait rendre le contrôle du diabète plus difficile. Il faut savoir que la biodisponibilité de la metformine à libération non retardée, à contratio de celle de la metformine à libération retardée, est réduite lors d'une prise avec de la nourriture.
Interactions
Sitagliptine et metformine
Chez les patients diabétiques de type 2, un traitement associé avec des doses multiples de sitagliptine (50 mg 2x par jour) et de metformine (1000 mg 2x par jour) n'a influencé significativement ni la pharmacocinétique de la sitagliptine ni celle de la metformine.
Aucune étude d'interaction pharmacocinétique n'a été effectuée sur Janumet XR. Toutefois, de telles études ont été réalisées sur les principes actifs individuels de Janumet XR, c'est-à-dire sur la sitagliptine et sur la metformine.
Phosphate de sitagliptine
Des études d'interaction pharmacologique avec la sitagliptine n'ont montré aucune influence cliniquement significative sur la pharmacocinétique des médicaments suivants: metformine, rosiglitazone, glibenclamide, simvastatine, warfarine et contraceptifs oraux. Ces données laissent supposer que la sitagliptine n'inhibe pas les isoenzymes CYP3A4, 2C8 et 2C9. Des données obtenues in vitro laissent également supposer que la sitagliptine n'inhibe pas les CYP2D6, 1A2, 2C19 ou 2B6 et n'induit pas le CYP3A4.
Des analyses pharmacocinétiques de population auprès de patients diabétiques de type 2 n'ont constaté aucune influence significative de la médication concomitante sur la pharmacocinétique de la sitagliptine. Ces analyses se sont penchées sur les médicaments usuels utilisés chez les patients diabétiques de type 2.
L'administration simultanée de sitagliptine et de digoxine a provoqué une légère augmentation de l'aire sous la courbe (AUC, 11%) ainsi que de la concentration plasmatique maximale moyenne (Cmax, 18%) de la digoxine. Ces modifications ne sont pas cliniquement significatives. Les patients prenant de la digoxine devraient être surveillés en conséquence.
L'administration simultanée d'une dose orale unique de 100 mg de sitagliptine et d'une dose unique orale de 600 mg de ciclosporine, un inhibiteur puissant de la glycoprotéine-p, a provoqué une augmentation de l'AUC de la sitagliptine de près de 29% ainsi qu'une hausse de la Cmax de la sitagliptine de 68%. Les modifications observées au niveau de la pharmacocinétique de la sitagliptine ne sont pas considérées comme cliniquement significatives.
Chlorhydrate de metformine
Glibenclamide: Dans une étude d'interaction avec administration unique à des patients diabétiques de type 2, la prise simultanée de metformine et de glibenclamide n'a modifié ni la pharmacocinétique ni la pharmacodynamie de la metformine.
Des valeurs réduites de l'AUC et de la Cmax du glibenclamide ont été observées mais de façon fortement variable. Vu l'administration unique des médicaments dans cette étude et vu l'absence d'une corrélation entre le taux sanguin de glibenclamide et les effets pharmacodynamiques, il est impossible de se prononcer clairement sur la signification clinique de cette interaction.
Furosémide: Une étude d'interaction avec administration unique de metformine et de furosémide chez des volontaires sains a montré que les paramètres pharmacocinétiques des deux principes actifs ont été influencés. Le furosémide a fait augmenter la Cmax de la metformine de 22% dans le plasma et le sang et l'AUC de la metformine de 15% dans le sang, sans exercer une influence significative sur la clairance rénale de la metformine. Lors d'une administration concomitante de metformine, la Cmax et l'AUC du furosémide ont été réduites respectivement de 31% et de 12% par rapport à la monothérapie. La demi-vie terminale a été raccourcie de 32% sans influence significative sur la clairance rénale du furosémide. On ne dispose pas de données sur l'interaction entre metformine et furosémide dans le cadre d'un traitement prolongé.
Nifédipine: Une étude d'interaction avec administration unique de metformine et de nifédipine chez des volontaires sains a montré que l'administration concomitante de nifédipine faisait augmenter la Cmax et l'AUC de la metformine respectivement de 20% et de 9% dans le plasma et que la metformine était davantage éliminée dans l'urine.
Les valeurs de Tmax et de demi-vie n'ont pas été influencées. La nifédipine semble accélérer l'absorption de la metformine. La metformine n'exerce guère d'influence sur la nifédipine.
L'administration concomitante de médicaments qui interfèrent avec les systèmes de transport tubulaires rénaux impliqués dans l'élimination rénale de la metformine (par ex. le transporteur de cations organiques 2 [OCT2] / les inhibiteurs des transporteurs d'extrusion de multiples médicaments et toxines [MATE] tels que la ranolazine, le vandétanib, le dolutégravir et la cimétidine), pourrait augmenter l'exposition systémique de la metformine et, par conséquent, le risque d'une acidose lactique. Dans des études d'interaction avec administration unique et répétée de metformine en association avec la cimétidine à des sujets sains, les concentrations maximales de metformine dans le plasma et le sang complet ont augmenté de 60% et l'AUC plasmatique et l'AUC pour le sang complet de la metformine ont augmenté de 40%. Le bénéfice et les risques de l'utilisation concomitante de ces substances avec la metformine doivent donc être soigneusement évalués.
Autres: Certains médicaments peuvent déclencher une hyperglycémie et perturber ainsi l'équilibre glycémique. Ces médicaments englobent par exemple les diurétiques thiazidiques et autres, les corticostéroïdes, les phénothiazines, les médicaments pour la glande thyroïde, les œstrogènes, les contraceptifs oraux, la phénytoïne, la niacine, les sympathicomimétiques, les bloqueurs des canaux calciques et l'isoniazide. Si l'un de ces médicaments est administré à un patient traité par Janumet XR, ce patient doit être surveillé de près pour assurer un équilibre glycémique adéquat.
Dans des études d'interaction avec administration unique chez des volontaires sains, la pharmacocinétique de la metformine et du propranolol ainsi que la pharmacocinétique de la metformine et de l'ibuprofène n'ont pas été influencées lors d'une administration des associations correspondantes.
La metformine ne se lie presque pas aux protéines plasmatiques et n'interagit donc guère – contrairement aux sulfonylurées, qui se lient fortement aux protéines sériques – avec les médicaments fortement liés aux protéines, par exemple les salicylates, les sulfamides, le chloramphénicol et le probénécide.
Grossesse/Allaitement
Emploi pendant la grossesse
Aucune étude adéquate et sévèrement contrôlée n'a été réalisée chez les femmes enceintes sur Janumet XR ou ses composants individuels. C'est pourquoi la sécurité de Janumet XR chez les femmes enceintes n'est pas connue.
La prise de Janumet XR, ainsi que celle d'autres antidiabétiques oraux, n'est pas recommandée durant la grossesse.
Emploi en période d'allaitement
Aucune étude n'a été effectuée sur des animaux en lactation avec les principes actifs associés de Janumet XR. Dans les études sur les principes actifs individuels, aussi bien la sitagliptine que la metformine ont été excrétées dans le lait des rates en lactation. On ignore si la sitagliptine passe également dans le lait maternel humain. C'est pourquoi Janumet XR ne devrait pas être utilisé en période d'allaitement.
Effet sur l’aptitude à la conduite et l’utilisation de machines
Aucune étude concernant les effets de Janumet XR sur l'aptitude à conduire et à utiliser des machines n'a été menée. On ne s'attend toutefois pas à ce que Janumet XR influence les aptitudes à la conduite et l'utilisation de machines.
Effets indésirables
Aucune étude clinique n'a été effectuée sur les comprimés Janumet XR. Par contre, la bioéquivalence entre Janumet XR et une co-administration de sitagliptine et de metformine à libération retardée a été démontrée (voir «Pharmacocinétique»).
Dans les études cliniques contrôlées par placebo, le traitement associé à la sitagliptine et à la metformine à libération non retardée a généralement été bien toléré par les patients souffrant d'un diabète de type 2. L'incidence globale des effets indésirables chez les patients sous traitement associé à la sitagliptine et à la metformine a été similaire à celle observée sous placebo et metformine.
Les incidences des effets indésirables sont définies comme suit: fréquents ≥1/100, <1/10; occasionnels ≥1/1000, <1/100; rares ≥1/10000, <1/1000; très rares <1/10000.
Traitement d'association à la sitagliptine et à la metformine
Une étude de 24 semaines avec contrôle par placebo, effectuée auprès de patients chez lesquels un régime alimentaire et un exercice physique régulier n'avaient pas permis d'obtenir un équilibre glycémique suffisant, a examiné le traitement par la sitagliptine 50 mg 2x par jour en association avec la metformine 500 ou 1000 mg 2x par jour. Les effets indésirables en rapport avec le traitement associé qui se sont produits chez ≥1% des patients (et ont été plus fréquents que dans le groupe recevant le placebo) ont été les suivants:
Troubles du métabolisme et de la nutrition:
Fréquents: hypoglycémie.
Troubles du système nerveux
Fréquents: céphalées.
Troubles gastro-intestinaux
Fréquents: diarrhées, nausées, dyspepsie, ballonnements, vomissements.
Dans une étude de 24 semaines avec contrôle par placebo, examinant la sitagliptine administrée en plus d'un traitement déjà commencé à la metformine, 464 patients traités à la metformine ont reçu 100 mg de sitagliptine une fois par jour, tandis que 237 autres patients traités à la metformine ont reçu un placebo.
Les effets indésirables en rapport avec le traitement associé qui se sont produits chez ≥1% des patients (et ont été plus fréquents que dans le groupe recevant le placebo) ont été les suivants:
Troubles gastro-intestinaux
Fréquents: nausées.
Hypoglycémie et troubles gastro-intestinaux
Dans les études contrôlées par placebo sur la sitagliptine et la metformine en tant que traitement associé, l'incidence des hypoglycémies (indépendamment de l'évaluation de la causalité par le médecin investigateur) dans le cadre d'une prise de sitagliptine en association avec de la metformine a été comparable à celle sous metformine plus placebo.
L'incidence des troubles gastro-intestinaux chez les patients traités par l'association sitagliptine-metformine a été comparable à celle chez les patients sous metformine seule (voir le Tableau 1).
Tableau 1:
Hypoglycémie et troubles gastro-intestinaux (indépendamment de l'évaluation de la causalité par le médecin investigateur) chez les patients sous traitement associé
Nombre de patients (%) | ||||||
Étude sur la sitagliptine et la metformine chez des patients dont l'équilibre glycémique était insuffisant avec un régime alimentaire et un exercice physique régulier comme seuls traitements | Étude sur la sitagliptine en tant que traitement complémentaire en plus de la metformine | |||||
Placebo | Sitagliptine | Metformine | Sitagliptine | Placebo et metformine | Sitagliptine 100 mg 1x par jour et metformine | |
| N= 176 | N= 179 | N= 364 | N= 372 | N= 237 | N= 464 | |
| Hypoglycémie | 1 (0.6) | 1 (0.6) | 3 (0.8) | 6 (1.6) | 5 (2.1) | 6 (1.3) |
| Diarrhées | 7 (4.0) | 5 (2.8) | 28 (7.7) | 28 (7.5) | 6 (2.5) | 11 (2.4) |
| Nausées | 2 (1.1) | 2 (1.1) | 20 (5.5) | 18 (4.8) | 2 (0.8) | 6 (1.3) |
| Vomissements | 1 (0.6) | 0 (0.0) | 2 (0.5) | 8 (2.1) | 2 (0.8) | 5 (1.1) |
| Douleurs abdominales† | 4 (2.3) | 6 (3.4) | 14 (3.8) | 11(3.0) | 9 (3.8) | 10 (2.2) |
† Dans l'étude auprès de patients insuffisamment contrôlés par un régime alimentaire et un exercice physique régulier, les symptômes abdominaux ont également été comptés en tant que douleurs abdominales.
†† Données groupées pour les patients ayant reçu de plus faibles / de plus fortes doses de metformine.
Dans toutes les études, tous les effets indésirables considérés comme des épisodes d'hypoglycémie – avec ou sans confirmation par un contrôle de la glycémie – ont été pris en compte.
Sitagliptine en association avec la metformine et une sulfonylurée
Dans une étude de 24 semaines, effectuée avec contrôle par placebo, les participants ont reçu une dose quotidienne de 100 mg de sitagliptine en plus d'un traitement déjà en cours avec des doses quotidiennes de ≥4 mg de glimépiride et de ≥1500 mg de metformine. Les effets indésirables médicamenteux suivants ont été observés chez ≥1% des patients sous sitagliptine (n= 116) et plus fréquemment que chez les patients sous placebo (n= 113):
Troubles du métabolisme et de la nutrition
Fréquents: hypoglycémie.
Troubles gastro-intestinaux
Fréquents: constipation.
Aucune altération cliniquement significative des fonctions vitales ou de l'ECG (y compris intervalle QT) n'a été observée pendant le traitement à la sitagliptine associée à la metformine.
Sitagliptine en association avec la metformine et l'insuline
Dans une étude de 24 semaines avec contrôle par placebo, examinant la sitagliptine 100 mg ajoutée à un traitement en cours associant la metformine ≥1500 mg par jour et une dose stable d'insuline, l'hypoglycémie a été la seule réaction indésirable observée sous sitagliptine chez ≥1% des patients (n= 229) et plus souvent que sous placebo (n= 233) (sitagliptine 15,3%, placebo 8,2%). Ces incidences correspondent à tous les effets indésirables indépendamment de la présence ou non d'un rapport de causalité selon l'appréciation du médecin investigateur. En particulier pour les taux d'hypoglycémie, il faut considérer que tous les patients étaient traités non seulement avec la sitagliptine ou avec le placebo, mais aussi avec l'insuline et dans certains cas également avec la metformine. Dans une autre étude de 24 semaines chez des patients qui, pendant une intensification de l'insuline (avec ou sans metformine) ont reçu de la sitagliptine en traitement complémentaire, les effets indésirables qui ont été observés chez ≥1% des patients traités par sitagliptine et metformine (N= 285) et qui étaient plus fréquents que chez les patients sous placebo et metformine (N= 283), étaient: constipation (sitagliptine et metformine, 1,4%; placebo et metformine, 1,1%), diarrhée (4,9%; 2,5%), vomissements (3,2%; 1,1%), œdème périphérique (2,1%; 1,4%), fièvre (1,1%; 0,4%), bronchite (1,4%; 1,1%), cellulite (1,4%; 1,1%), pharyngite (1,8%; 1,1%), infection des voies respiratoires supérieures (4,2%; 1,4%), diminution de la clairance de la créatinine (1,1%; 0,0%), douleurs musculo-squelettiques (1,4%; 1,1%), myalgie (1,1%; 0,7%), néphrolithiase (1,1%; 0,4%) et toux (2,8%; 1,8%). Ces incidences correspondent à tous les effets indésirables indépendamment de la présence ou non d'un rapport de causalité selon l'appréciation des médecins investigateurs. De plus, l'incidence d'hypoglycémie était de 24,9% chez les patients traités par sitagliptine, metformine et insuline et de 37,8% chez les patients sous placebo, metformine et insuline.
Effets indésirables connus sous sitagliptine
Chez les patients prenant de la sitagliptine, aucun effet indésirable médicamenteux n'a été observé à une incidence ≥1%.
Effets indésirables connus sous metformine
Troubles de la circulation sanguine et lymphatique
Cas isolés de leucopénie, de thrombopénie et d'anémie hémolytique.
Très rares: taux sanguin réduit de vitamine B12.
Troubles du métabolisme et de la nutrition
Très rares: acidose lactique (incidence: 3 cas/100'000 patient-années, cf. la section «Mises en garde et précautions»).
Troubles du système nerveux
Fréquents: goût métallique dans la bouche (3%).
Troubles gastro-intestinaux
Fréquents: troubles gastro-intestinaux tels que nausées, vomissements, diarrhées, douleurs abdominales, perte d'appétit.
Ces symptômes se manifestent généralement au début du traitement et régressent en général spontanément par la suite.
Troubles hépato-biliaires
Dans des cas isolés: valeurs anormales des tests hépatiques fonctionnels, par exemple taux accrus de transaminases ou hépatite (réversible après l'arrêt d'administration de la metformine).
Troubles cutanés et des tissus sous-cutanés
Très rares: réactions cutanées telles qu'érythème, prurit, urticaire.
Étude de sécurité cardiovasculaire TECOS: L'étude TECOS (Trial Evaluating Cardiovascular Outcomes with Sitagliptin) incluait une population en intention de traiter dont 7332 patients traités par 100 mg de sitagliptine par jour (ou 50 mg par jour si le débit de filtration glomérulaire estimé [DFGe] se situait entre ≥30 et <50 ml/min/1,73 m2 au début de l'étude) et 7339 patients traités par placebo. Les deux traitements étaient ajoutés au traitement conventionnel. La population étudiée comprenait au total 2004 patients âgés de ≥75 ans (970 traités par la sitagliptine et 1034 par placebo). La fréquence des événements indésirables sévères était généralement comparable chez les patients sous sitagliptine et ceux sous placebo. Une évaluation des complications liées au diabète prédéfinies a révélé des incidences semblables entre les groupes, y compris au niveau des infections (18,4% des patients traités par la sitagliptine et 17,7% des patients traités par placebo) et de l'insuffisance rénale (1,4% des patients traités par la sitagliptine et 1,5% des patients traités par placebo). Le profil des événements indésirables chez les patients âgés de ≥75 ans était généralement similaire à celui de la population totale.
Chez les patients recevant de l'insuline et/ou une sulfonylurée au début de l'étude, l'incidence d'une hypoglycémie sévère était de 2,7% chez les patients traités par la sitagliptine et de 2,5% chez les patients traités par placebo. Chez les patients n' utilisant pas d'insuline et/ou pas de sulfonylurée au début de l'étude, l'incidence d'une hypoglycémie sévère était de 1,0% chez les patients traités par la sitagliptine et de 0,7% chez les patients traités par placebo. L'incidence des pancréatites confirmées par un comité de vérification était de 0,3% chez les patients traités par la sitagliptine et de 0,2% chez les patients traités par placebo. L'incidence des tumeurs malignes confirmées par un comité de vérification était de 3,7% chez les patients traités par la sitagliptine et de 4,0% chez ceux traités par placebo.
Pancréatite: Dans une analyse cumulée de 19 études en double aveugle totalisant 10'246 patients randomisés sous sitagliptine 100 mg par jour (n = 5429) ou sous un traitement de contrôle correspondant (agent actif ou placebo) (n = 4817), l'incidence de pancréatite aiguë a été de 0,1 sur 100 patients-années dans tous les groupes (4 patients avec 1 événement sur 4708 patients-années sous sitagliptine, 4 patients avec 1 événement sur 3942 patients-années sous traitement de contrôle) (voir aussi Étude de sécurité cardiovasculaire TECOS et «Mises en garde et précautions», Pancréatite).
Expériences post-commercialisation
Des réactions indésirables additionnelles ont été signalées après la commercialisation de Janumet XR ou de la sitagliptine, l'un des composants de Janumet XR. Ces effets indésirables ont été rapportés lors de l'utilisation de Janumet XR ou de sitagliptine, seuls et/ou en association avec d'autres substances anti-hyperglycémiantes. Vu que ces données reposent sur des rapports spontanés issus d'une population de taille inconnue, il est impossible d'en déterminer l'incidence ou la causalité: réactions d'hypersensibilité, y compris anaphylaxie, angio-œdème, éruption cutanée, urticaire, vasculite cutanée et anomalies cutanées exfoliatives, y compris syndrome de Stevens-Johnson (voir les sections «Contre-indications» et «Mises en garde et précautions», Réactions d'hypersensibilité); pancréatite aiguë, y compris pancréatite hémorragique avec issue fatale et non fatale et pancréatite nécrosante (voir aussi «Mises en garde et précautions»); détérioration de la fonction rénale, y compris insuffisance rénale aiguë (dans certains cas, une dialyse s'avère nécessaire); rhabdomyolyse; pemphigoïde bulleuse (voir «Mises en garde et précautions», Pemphigoïde bulleuse); infections des voies respiratoires supérieures, nasopharyngite; constipation, vomissements; céphalées; arthralgie, myalgie, douleur des extrémités, douleur dorsale; prurit.
Investigations
Phosphate de sitagliptine
L'incidence des anomalies de paramètres de laboratoire a été similaire entre le groupe sous sitagliptine et metformine et le groupe témoin sous placebo et metformine.
Plusieurs études cliniques ont constaté une légère augmentation du taux de leucocytes (environ 200 cellules par µl par rapport au placebo; valeur initiale moyenne de leucocytes: environ 6600 cellules par µl), en raison d'une légère augmentation du nombre de neutrophiles. Cette observation a été faite dans la plupart des études, mais non dans toutes. Ces anomalies des paramètres de laboratoire ont été jugées sans importance clinique.
Chlorhydrate de metformine
Dans des études cliniques contrôlées sur l'administration de metformine pendant 29 semaines, environ 7% des patients ont présenté une réduction du taux de vitamine B12 à des valeurs inférieures à la moyenne alors que les valeurs initiales avaient été normales. Toutefois, cette réduction n'a pas été associée à des manifestations cliniques.
Une telle réduction du taux sanguin de vitamine B12, probablement due à une interférence avec l'absorption de la vitamine B12 par le complexe B12-facteur intrinsèque, n'est cependant que très rarement associée à une anémie et semble être rapidement réversible dès que l'administration de metformine est arrêtée ou que des suppléments de vitamine B12 sont administrés (voir la section «Mises en garde et précautions», Chlorhydrate de metformine).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Phosphate de sitagliptine
Dans les études cliniques contrôlées auprès de volontaires sains, des doses uniques de sitagliptine atteignant jusqu'à 800 mg ont généralement été bien tolérées.
Pour une dose de 800 mg de sitagliptine, une étude a observé des sus-décalages minimaux de l'intervalle QT, cependant jugés comme sans signification clinique.
Dans les études cliniques il n'existe pas de données sur les doses supérieures à 800 mg.
Dans des études de phase I avec administrations multiples de sitagliptine à des doses allant jusqu'à 600 mg par jour pendant ≤10 jours et à des doses allant jusqu'à 400 mg par jour pendant des périodes couvrant jusqu'à 28 jours, aucun effet indésirable dépendant de la dose n'a été observé cliniquement.
Dans le cas d'un surdosage, il convient de prendre les mesures de soutien habituelles, par exemple l'élimination des substances actives encore non absorbées dans le tractus gastro-intestinal, la surveillance clinique (y compris par ECG) et, si nécessaire, l'instauration d'un traitement de soutien.
La sitagliptine n'est que modérément dialysable. Au cours d'études cliniques, près de 13,5% de la dose a été éliminée durant une hémodialyse de trois à quatre heures. Une hémodialyse plus longue peut être envisagée si elle est cliniquement utile. On ignore si la sitagliptine peut être dialysée par dialyse péritonéale.
Chlorhydrate de metformine
Des surdosages de metformine se sont déjà produits, même à des doses de plus de 50 grammes. Une hypoglycémie a été observée dans environ 10% des cas, cependant sans qu'un rapport causal avec la metformine ait pu être démontré. Dans environ 32% des cas de surdosage à la metformine, une acidose lactique a été constatée (voir la section «Mises en garde et précautions», Chlorhydrate de metformine). La metformine étant dialysable (avec une clairance allant jusqu'à 170 ml/min dans de bonnes conditions hémodynamiques), une hémodialyse est recommandée pour éliminer la metformine accumulée.
Propriétés/Effets
Code ATC
A10BD07
Mécanisme d'action
Le phosphate de sitagliptine appartient à une classe d'antidiabétiques oraux, les inhibiteurs de la dipeptidylpeptidase-4 (DPP-4), qui améliorent le contrôle de la glycémie chez les patients atteints de diabète de type 2 en augmentant les concentrations des incrétines actives. Les incrétines, y compris le GLP-1 (Glucagon-like Peptide-1) et le GIP (Glucose-dependent insulinotropic Peptide), sont libérées toute la journée à partir des intestins. Leur concentration augmente lors de la prise de nourriture. Les incrétines font partie d'un système endogène qui intervient dans la régulation physiologique de l'homéostasie du glucose. Lorsque la glycémie est normale ou élevée, le GLP-1 et le GIP favorisent la synthèse d'insuline et la sécrétion d'insuline à partir des cellules bêta du pancréas par des signaux intracellulaires impliquant l'AMP cyclique. Le traitement du diabète de type 2 au GLP-1 ou aux inhibiteurs de la DPP-4 dans des modèles animaux a montré que la sensibilité des cellules bêta au glucose est améliorée et que la biosynthèse ainsi que la sécrétion d'insuline sont stimulées. En présence de concentrations plus élevées d'insuline, l'absorption tissulaire de glucose augmente. Parallèlement, le GLP-1 diminue la sécrétion de glucagon à partir des cellules alpha du pancréas. De faibles concentrations de glucagon accompagnées de concentrations d'insuline élevées diminuent la production de glucose dans le foie, ce qui entraîne à son tour une baisse de la glycémie. Les effets du GLP-1 et du GIP sont dépendants du glucose. Lors d'une glycémie basse, le GLP-1 ne stimule ni la sécrétion d'insuline ni la suppression de la sécrétion de glucagon. Grâce au GLP-1 et au GIP, la sécrétion d'insuline est augmentée lorsque la glycémie dépasse les valeurs normales. Le GLP-1 n'affecte pas la réaction normale du glucagon lors d'hypoglycémie. L'activité du GLP-1 et du GIP est régulée par l'enzyme DPP-4, qui hydrolyse rapidement les incrétines et les transforme en substances inactives. La sitagliptine empêche l'hydrolyse des incrétines par la DPP-4, ce qui augmente les concentrations plasmatiques des formes actives du GLP-1 et du GIP. Suite à l'augmentation de la concentration d'incrétines actives, la sitagliptine provoque une hausse de la sécrétion d'insuline et une baisse de la concentration de glucagon, en fonction de la concentration de glucose. Chez les patients atteints de diabète de type 2 et présentant une hyperglycémie, ces modifications de la concentration d'insuline et de glucagon entraînent une baisse de la concentration de l'hémoglobine A1c (HbA1c) ainsi qu'une diminution des glycémies postprandiales et à jeun. Ce mécanisme dépendant du glucose se distingue de celui des sulfonylurées. Celles-ci provoquent une sécrétion d'insuline même en présence d'une glycémie basse, ce qui peut provoquer une hypoglycémie chez les patients diabétiques de type 2 et les sujets sains. Alors que la sitagliptine est un inhibiteur puissant et très sélectif de l'enzyme DPP-4, ses concentrations thérapeutiques n'inhibent pas les enzymes apparentées DPP-8 et DPP-9, très similaires.
Chlorhydrate de metformine
La metformine est un antidiabétique qui améliore la tolérance au glucose chez les patients souffrant de diabète de type 2 et réduit aussi bien la glycémie basale que la glycémie postprandiale. Les mécanismes d'action pharmacologiques de la metformine se distinguent de ceux des autres classes d'antidiabétiques oraux. La metformine réduit aussi bien la synthèse hépatique de glucose que l'absorption intestinale de glucose; elle améliore la sensibilité à l'insuline en augmentant l'assimilation périphérique et l'utilisation du glucose. Contrairement aux sulfonylurées, la metformine ne provoque pas d'hypoglycémie chez les patients diabétiques de type 2 ou chez les sujets sains, sauf dans des cas très particuliers (voir la section «Mises en garde et précautions», Chlorhydrate de metformine). Elle ne provoque pas non plus d'hyperinsulinémie. Pendant un traitement à la metformine, la sécrétion d'insuline reste inchangée tandis que les taux d'insuline à jeun et la réponse insulinique dans le plasma au cours de la journée peuvent même baisser.
Pharmacodynamique
Non pertinent.
Efficacité clinique
Études cliniques
Aucune étude clinique n'a été effectuée sur l'efficacité et la sécurité de Janumet XR. Des études cliniques avec administration concomitante de sitagliptine et de metformine à libération non retardée ont constaté une amélioration significative du contrôle de l'équilibre glycémique chez les patients souffrant d'un diabète de type 2. La bioéquivalence entre Janumet XR et un traitement consistant en une administration concomitante de sitagliptine et de comprimés de metformine à libération retardée a été démontrée pour toutes les puissances de dosage.
Dans une étude comparative, l'administration une fois par jour de metformine à libération retardée a permis pour tous les paramètres glycémiques évalués une efficacité comparable à celle de l'administration usuellement prescrite de metformine à libération non retardée deux fois par jour.
Sitagliptine et metformine à libération non retardée chez les patients diabétiques de type 2
Une étude en double aveugle de 24 semaines, randomisée, avec contrôle par placebo, a examiné la sécurité et l'efficacité de l'association de sitagliptine et de metformine chez 1091 patients diabétiques de type 2 qui présentaient un équilibre glycémique insuffisant malgré un régime alimentaire et un exercice physique régulier.
Chez les patients ayant un contrôle glycémique insuffisant sous régime alimentaire plus exercice physique, l'association sitagliptine-metformine a permis des améliorations significatives en ce qui concerne le taux d'HbA1c, le taux plasmatique de glucose à jeun (FPG) et la valeur postprandiale à 2 h du taux plasmatique de glucose (PPG) par rapport au placebo, à la metformine seule et à la sitagliptine seule (p <0,001; Tableau 2, Figure 1). Une amélioration avec réduction presque maximale du FPG a été atteinte au premier bilan à 3 semaines (correspondant au premier bilan après le début du traitement) et a pu être maintenue pendant toute la durée de cette étude de 24 semaines. En comparaison avec le groupe sous placebo, les patients qui avaient eu des valeurs initiales plus élevées d'HbA1c ont bénéficié d'une réduction moyenne plus importante du taux d'HbA1c.
L'amélioration du taux d'HbA1c n'a pas été influencée par le sexe, l'âge, la race ou la valeur initiale du BMI. Les examens de la fonction des cellules bêta, HOMA-β, de même que le rapport pro-insuline/insuline, ont également montré une amélioration sous l'association sitagliptine-metformine en comparaison avec chacune des substances seules. Les paramètres lipidiques n'ont pas été influencés. Les réductions moyennes du taux d'HbA1c observées dans le sous-groupe des patients sans traitement antidiabétique avant l'étude ont été: -1,06% sous sitagliptine 100 mg une fois par jour (n= 88); -1,09% sous metformine 500 mg deux fois par jour (n= 90); -1,24% sous metformine 1000 mg deux fois par jour (n= 87); -1,59% sous sitagliptine 50 mg deux fois par jour et metformine 500 mg deux fois par jour (n= 100); -1,94% sous sitagliptine 50 mg deux fois par jour et metformine 1000 mg deux fois par jour (n= 86); -0,17% sous placebo (n= 83).
Fig. 1: Variation moyenne du taux d'HbA1c sur 24 semaines par rapport aux valeurs initiales, sous sitagliptine seule, metformine seule et sous association des deux substances chez des patients diabétiques de type 2†
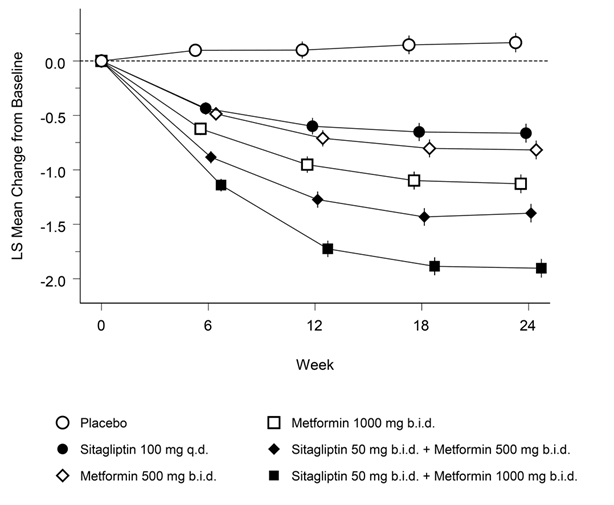
† Population de tous les patients traités (analyse en intention de traitement, ITT), moyennes des moindres carrés, ajustées en fonction du traitement antidiabétique antérieur et de la valeur initiale.
Tableau 2:
Paramètres glycémiques et poids corporel lors de l'examen final à 24 semaines†
Placebo | Sitagliptine | Metformine | Sitagliptine | Metformine | Sitagliptine | |
HbA1c (%) | N= 165 | N= 175 | N= 178 | N= 183 | N= 177 | N= 178 |
Valeur initiale (moyenne) | 8,68 | 8,87 | 8,90 | 8,79 | 8,68 | 8,76 |
Modification par rapport à la valeur initiale (moyenne ajustée‡) | 0,17 | -0,66 | -0,82 | -1,40 | -1,13 | -1,90 |
Différence par rapport au placebo (moyenne ajustée‡) | - | -0,83§ | -0,99§ | -1,57§ | -1,30§ | -2,07§ |
Patients (%) avec taux d'HbA1c <7% | 15 (9,1) | 35 (20,0) | 41 (23,0) | 79 (43,2) | 68 (38,4) | 118 (66,3) |
FPG (mmol/l) | N= 169 | N= 178 | N= 179 | N= 183 | N= 179 | N= 180 |
Valeur initiale (moyenne) | 10,91 | 11,19 | 11,40 | 11,33 | 10,94 | 10,93 |
Modification par rapport à la valeur initiale (moyenne ajustée‡) | 0,32 | -0,97 | -1,52 | -2,62 | -1,63 | -3,55 |
Différence par rapport au placebo (moyenne ajustée‡) | - | -1,29§ | -1,84§ | -2,94§ | -1,95§ | -3,87§ |
PPG à 2 h (mmol/l) | N= 129 | N= 136 | N= 141 | N= 147 | N= 138 | N= 152 |
Valeur initiale (moyenne) | 15,38 | 15,86 | 16,26 | 16,21 | 15,74 | 15,94 |
Modification par rapport à la valeur initiale (moyenne ajustée‡) | 0,02 | -2,88 | -2,97 | -5,14 | -4,33 | -6,48 |
Différence par rapport au placebo (moyenne ajustée‡) | -2,9§ | -2,98§ | -5,16§ | -4,35§ | -6,49§ | |
Poids corporel (kg)** | N= 167 | N= 175 | N= 179 | N= 184 | N= 175 | N= 178 |
Valeur initiale (moyenne) | 90,1 | 85,9 | 88,1 | 90,0 | 89,4 | 88,2 |
Modification par rapport à la valeur initiale (moyenne ajustée‡) | -0,9 | 0,0 | -0,9 | -0,6 | -1,1 | -1,3 |
Différence par rapport au placebo (moyenne ajustée‡) | 0,9¶ | 0,1# | 0,4# | -0,1# | -0,3# |
† All Patients Treated Population (une analyse en intention de traitement, ITT).
‡ Moyennes des moindres carrés, ajustées en fonction du traitement antidiabétique antérieur et de la valeur initiale.
§ p <0,001 par rapport au placebo.
** Population de tous les patients tels que traités (all Patients as Treated, APaT) à l'exclusion des données après un traitement glycémique d'urgence.
¶ p = 0,005 par rapport au placebo.
# Statistiquement non significatif (p ≥0,05) par rapport au placebo.
En outre, cette étude a inclus des patients (n = 117) présentant une forte hyperglycémie (HbA1c >11% ou glycémie >15,56 mmol/l), traités deux fois par jour à la sitagliptine 50 mg et à la metformine 1000 mg. Aucun contrôle avec placebo n'a été fait pour ce groupe de patients. Dans ce groupe de patients, le taux initial moyen d'HbA1c était de 11,15%, le FPG de 17,47 mmol et la valeur de PPG à deux heures de 24,5 mmol/l. Au bout de 24 semaines, les réductions suivantes ont été constatées par rapport aux valeurs initiales: ‑2,9% pour le taux d'HbA1c, ‑7,04 mmol/l pour le FPG et –11,55 mmol/l pour le PPG à 2 heures. Dans cette cohorte, une augmentation de 1,3 kg du poids corporel a été constatée à 24 semaines.
Traitement complémentaire à la sitagliptine chez les patients n'atteignant pas un bon équilibre glycémique sous metformine seule
Deux études cliniques effectuées en double aveugle avec contrôle par placebo auprès de patients diabétiques de type 2 ont évalué la sécurité et l'efficacité d'une association sitagliptine-metformine à libération non retardée. Dans les deux études, des patients présentant une glycémie mal contrôlée ont poursuivi leurs prises de metformine ≥1500 mg tout en prenant de façon randomisée soit 100 mg de sitagliptine, soit un placebo en plus du traitement en cours à la metformine.
Dans une étude, 701 patients ont été traités 24 semaines soit avec 100 mg de sitagliptine, soit avec un placebo. L'ajout de la sitagliptine au traitement en cours à la metformine à libération non retardée a permis une amélioration significative du taux d'HbA1c (-0,65%), du FPG (-1,41 mmol/l) et de la glycémie postprandiale à 2 h (-2,81 mmol/l) en comparaison avec l'ajout du placebo au traitement en cours à la metformine (voir la Fig. 2 et Tableau 3). L'amélioration du taux d'HbA1C par rapport au placebo n'a pas été influencée par le taux initial d'HbA1C, les traitements antidiabétiques antérieurs, le sexe, l'âge, le BMI initial, le temps écoulé depuis le diagnostic du diabète, la présence d'un syndrome métabolique ou les indices standard de l'insulinorésistance (HOMA-IR) ou de la sécrétion d'insuline (HOMA-β).
Fig. 2: Modification moyenne du taux d'HbA1c à 24 semaines par rapport à la valeur initiale chez des patients diabétiques de type 2 traités avec une dose quotidienne de 100 mg de sitagliptine en association avec de la metformine ou avec un placebo en plus de la metformine† ‡
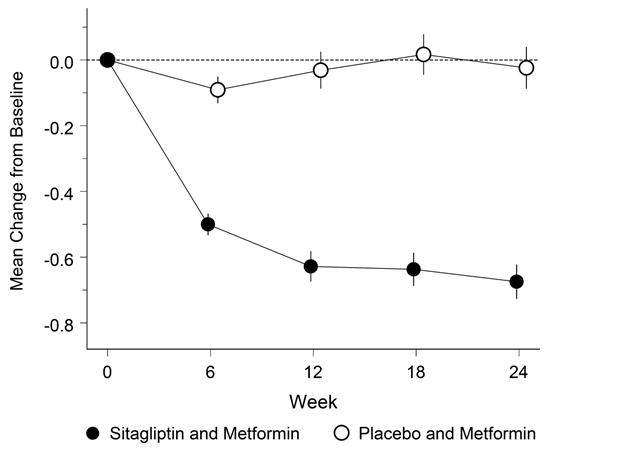
† Patients n'ayant pas atteint un équilibre glycémique suffisant sous metformine seule.
‡ All Patients Treated Population. Moyennes des moindres carrés, ajustées en fonction du traitement antidiabétique antérieur et de la valeur initiale.
Tableau 3:
Paramètres glycémiques et poids corporel lors de l'examen final (étude de 24 semaines)
Traitement complémentaire à la sitagliptine chez des patients n'ayant pas atteint un bon équilibre glycémique sous metformine seule†
Sitagliptine 100 mg par jour + metformine | Placebo + | |
HbA1c (%) | N= 453 | N= 224 |
Valeur initiale (moyenne) | 7,96 | 8,03 |
Modification par rapport à la valeur initiale (moyenne ajustée‡) | -0,67 | -0,02 |
Différence par rapport au placebo + metformine (moyenne ajustée ‡) | -0,65§ | |
Patients (%) avec HbA1c <7% | 213 (47,0) | 41 (18,3) |
FPG (mmol/l) | N= 454 | N= 226 |
Valeur initiale (moyenne) | 9,44 | 9,64 |
Modification par rapport à la valeur initiale (moyenne ajustée‡) | -0,94 | 0,47 |
Différence par rapport au placebo + metformine (moyenne ajustée‡) | -1,41§ | |
PPG à 2 h (mmol/l) | N= 387 | N= 182 |
Valeur initiale (moyenne) | 15,25 | 15,13 |
Modification par rapport à la valeur initiale (moyenne ajustée‡) | -3,44 | -0,63 |
Différence par rapport au placebo + metformine (moyenne ajustée‡) | -2,81§ | |
Poids corporel (kg)** | N= 399 | N= 169 |
Valeur initiale (moyenne) | 86,9 | 87,6 |
Modification par rapport à la valeur initiale (moyenne ajustée‡) | -0,7 | -0,6 |
Différence par rapport au placebo + metformine (moyenne ajustée‡) | -0,1¶ |
† All Patients treated Population (une analyse Intention-to-treat).
‡ Moyennes des moindres carrés, ajustées en fonction du traitement antidiabétique antérieur et de la valeur initiale.
§ p <0,001 par rapport au placebo + metformine.
** All Patients as Treated (APaT) Population, à l'exclusion des données après un traitement glycémique d'urgence.
¶ Statistiquement non significatif (p≥0,05) par rapport au placebo + metformine.
Fig. 3: Profils glycémiques de 24 h chez des patients diabétiques de type 2 au bout de 4 semaines de traitement à la sitagliptine 50 mg 2x par jour associée à la metformine ou au placebo associé à la metformine†
† Patients n'ayant pas atteint un équilibre glycémique suffisant sous metformine seule.
Ajout de sitagliptine au traitement de patients n'ayant pas atteint un équilibre glycémique suffisant avec l'association de metformine à libération non retardée et de glimépiride
Une étude randomisée de 24 semaines, effectuée en double aveugle avec contrôle par placebo auprès de 441 patients diabétiques de type 2, a évalué l'efficacité de la sitagliptine à la dose de 100 mg une fois par jour en association avec le glimépiride (en monothérapie ou en association avec la metformine). Dans cette étude, 220 patients suivaient déjà un traitement associant le glimépiride (≥4 mg par jour) et la metformine (≥1500 mg par jour).
L'association de sitagliptine, de glimépiride et de metformine a permis en comparaison avec le placebo une réduction significative du taux d'HbA1c par rapport aux valeurs initiales (-0,89%) et de la glycémie à jeun (FPG -1,15 mmol/l) (voir le Tableau 4). Les patients ayant présenté initialement un taux plus élevé d'HbA1c ont atteint une plus forte réduction moyenne du taux d'HbA1c par rapport au placebo.
Tableau 4:
Paramètres glycémiques* et poids corporel à l'examen final (étude de 24 semaines) chez les patients traités à la sitagliptine en plus du traitement associant le glimépiride et la metformine†
Sitagliptine 100 mg | Placebo | |
HbA1c (%) | N= 115 | N= 105 |
Valeur initiale (moyenne) | 8,27 | 8,28 |
Variation vs valeur initiale (moyenne ajustée‡) | -0,59 | 0,30 |
Variation vs placebo (moyenne ajustée‡) | -0,89§ | |
Patients (%) avec un taux d'HbA1c <7% | 26 (22,6) | 1 (1,0) |
FPG (mmol/l) | N= 115 | N= 109 |
Valeur initiale (moyenne) | 9,96 | 9,94 |
Variation vs valeur initiale (moyenne ajustée‡) | -0,43 | 0,72 |
Variation vs placebo (moyenne ajustée‡) | -1,15§ | |
Poids corporel (kg)** | N= 102 | N= 74 |
Valeur initiale (moyenne) | 86,5 | 84,6 |
Variation vs valeur initiale (moyenne ajustée‡) | 0,4 | -0,7 |
Variation vs placebo (moyenne ajustée‡) | 1,1†† |
* Paramètres glycémiques primaires et secondaires: HbA1c et glycémie à jeun
† All Patients treated Population (une analyse Intention-to-treat).
‡ Moyennes des moindres carrés, ajustées en fonction du traitement antérieur et de la valeur initiale.
§ p <0,001 versus placebo.
** All Patients as Treated (APaT) Population, à l'exclusion des données après un traitement glycémique d'urgence.
†† p = 0,007 versus placebo
Sitagliptine en tant que traitement complémentaire chez les patients insuffisamment traités avec une association de metformine à libération non retardée et d'insuline
641 patients diabétiques de type 2 ont participé à une étude randomisée de 24 semaines, réalisée en double aveugle avec contrôle par placebo, dans laquelle on a évalué l'efficacité de la sitagliptine 100 mg une fois par jour en association avec une dose stable d'insuline. Environ 75% des patients prenaient également de la metformine. Les patients sous insulinothérapie (insuline prête à l'emploi avec une durée d'action intermédiaire ou longue) en association ou non avec de la metformine ont été randomisés pour recevoir 100 mg de sitagliptine ou un placebo en tant que traitement complémentaire. Les critères glycémiques évalués ont inclus le taux de HbA1c, la glycémie à jeun et la glycémie postprandiale (PPG) au bout de 2 heures.
L'association de sitagliptine, de metformine et d'insuline a permis en comparaison avec le placebo une amélioration significative du taux de HbA1c, de la glycémie à jeun et de la PPG au bout de 2 heures (Tableau 5). L'amélioration du taux de HbA1c par rapport au placebo a été concordante dans tous les sous-groupes. Ces sous-groupes étaient définis en fonction de l'âge, du sexe, de la race, du BMI initial, de la durée du diabète depuis le diagnostic, de la présence ou non d'un syndrome métabolique et des indices standard d'insulinorésistance (HOMA-IR) et d'insulinosécrétion (HOMA-β). Une modification significative du poids corporel par rapport à la valeur initiale n'a été observée dans aucun des groupes.
Tableau 5:
Paramètres glycémiques et poids corporel lors de l'examen final (étude de 24 semaines) après la prise de sitagliptine en tant que traitement complémentaire en plus du traitement associant la metformine et une dose stable d'insuline†
Sitagliptine 100 mg | Placebo | |
HbA1c (%) | N= 223 | N= 229 |
Valeur initiale (moyenne) | 8,73 | 8,60 |
Modification par rapport à la valeur initiale (moyenne ajustée‡) | -0,66 | -0,13 |
Différence par rapport au placebo (moyenne ajustée‡,§) | -0,53** | |
Patients (%) avec HbA1c <7% | 32 (14,3) | 12 (5,2) |
FPG (mmol/l) | N= 225 | N= 229 |
Valeur initiale (moyenne) | 9,5 | 9,7 |
Modification par rapport à la valeur initiale (moyenne ajustée‡) | -1,2 | -0,2 |
Différence par rapport au placebo (moyenne ajustée‡) | -1,0** | |
PPG à 2 h (mmol/l) | N= 182 | N= 189 |
Valeur initiale (moyenne) | 15,4 | 15,4 |
Modification par rapport à la valeur initiale (moyenne ajustée‡) | -2,1 | 0,1 |
Différence par rapport au placebo (moyenne ajustée‡) | -2,2** | |
Poids corporel (kg)¶ | N= 201 | N= 200 |
Valeur initiale (moyenne) | 87,9 | 88,0 |
Modification par rapport à la valeur initiale (moyenne ajustée‡) | -0,1 | 0,0 |
Différence par rapport au placebo (moyenne ajustée‡) | 0,1# |
† All Patients treated Population (une analyse Intention-to-treat).
‡ Moyennes des moindres carrés, ajustées en fonction du traitement à l'insuline (prête à l'emploi ou non [de durée d'action intermédiaire ou longue]) lors de la visite 1 et en fonction de la valeur initiale.
§ Stratification en fonction de l'insuline; l'interaction n'était pas significative (p >0,10).
** p <0,001 versus placebo.
¶ All Patients as Treated (APaT) Population, à l'exclusion des données après un traitement glycémique d'urgence.
# Statistiquement non significatif (p ≥0,05) par rapport au placebo.
Dans une autre étude randomisée de 24 semaines, effectuée en double aveugle avec contrôle contre placebo, destinée à analyser l'effet de la sitagliptine en traitement concomitant complémentaire sur les économies d'insuline, 660 patients ayant un contrôle glycémique insuffisant sous insuline glargine avec ou sans metformine (≥1500 mg par jour) ont été randomisés en deux groupes: 100 mg sitagliptine (N= 330) ou placebo (N= 330). L'administration avait lieu une fois par jour parallèlement à une intensification du traitement par insuline. Chez les patients traités par metformine, le taux initial d'HbA1c était de 8,70% et la dose initiale d'insuline de 37 UI/jour. Les patients devaient titrer leur dose d'insuline glargine selon la glycémie à jeun obtenue après piqûre du doigt. Les critères d'évaluation secondaires de l'étude comprenaient l'HbA1c et la glycémie à jeun (FPG).
Chez les patients sous metformine, la hausse de la dose quotidienne d'insuline à semaine 24 était plus faible chez les patients traités par sitagliptine (19 UI/jour, N= 285) que chez les patients sous placebo (24 UI/jour, N= 283). Cette différence était statistiquement significative (p= 0,007).
Concernant les critères d'évaluation secondaires de l'étude, une réduction du taux d'HbA1c de -1,35% a été constatée chez les patients traités par sitagliptine, metformine et insuline contre -0,90% chez les patients sous placebo, metformine et insuline, soit une différence de ‑0,45% [IC à 95%: -0,62; -0,29]. La réduction du FPG chez les patients traités par sitagliptine, metformine et insuline était de -3,0 mmol/l contre -2,4 mmol/l chez les patients sous placebo, metformine et insuline, soit une différence de -0,7 mmol/l [IC à 95%: -1,0; ‑0,3].
Étude contrôlée avec substance active (glipizide) en association avec la metformine à libération non retardée
Les effets à long terme ont été examinés dans une étude randomisée de 52 semaines, effectuée en double aveugle avec contrôle avec glipizide auprès de patients diabétiques de type 2. Des patients n'ayant pas atteint un bon équilibre glycémique sous metformine (≥1500 mg par jour) ont reçu soit 100 mg de sitagliptine (n= 588), soit du glipizide (n= 584) pendant 52 semaines. Les patients sous glipizide ont reçu une dose initiale de 5 mg par jour, puis la dose a été augmentée progressivement de façon sélective au cours des 18 semaines suivantes sans provoquer d'hypoglycémie significative, jusqu'à l'obtention d'un glucose plasmatique à jeun de <110 mg/dl. Une dose maximale de 20 mg par jour était autorisée pour un équilibre glycémique optimal. Par la suite, la dose de glipizide a été maintenue constante. La dose moyenne de glipizide après l'adaptation initiale a été de 10,3 mg.
Les deux traitements ont conduit à une amélioration statistiquement significative de l'équilibre glycémique en comparaison avec les valeurs initiales. À 52 semaines, la réduction du taux d'HbA1c par rapport à la valeur initiale était de 0,67% chez les patients traités par 100 mg de sitagliptine par jour et de 0,67% chez les patients traités au glipizide. Ces résultats ont confirmé l'équivalence des deux principes actifs. La réduction de la glycémie à jeun a été de 0,56 mmol/l sous sitagliptine et de 0,42 mmol/l sous glipizide. Dans une analyse post hoc, les patients des deux groupes qui avaient présenté un taux initial plus élevé d'HbA1c (≥9%) ont bénéficié d'une réduction plus importante du taux d'HbA1c par rapport aux valeurs initiales (sitagliptine -1,68%; glipizide -1,76%). Le traitement à la sitagliptine a permis une réduction du rapport pro-insuline/insuline, un marqueur de la synthèse et de la sécrétion d'insuline. Sous glipizide, le rapport pro-insuline/insuline s'est aggravé. La fréquence des hypoglycémies a été significativement plus faible dans le groupe sous sitagliptine (4,9%) que dans le groupe sous glipizide (32,0%). Le poids corporel a baissé de façon significative chez les patients traités à la sitagliptine mais a augmenté significativement chez les patients traités au glipizide (-1,5 kg vs +1,1 kg).
Chlorhydrate de metformine
L'étude prospective randomisée UKPDS a confirmé les avantages à long terme d'un contrôle intensif de la glycémie dans le diabète de type 2. L'analyse des résultats – obtenus chez des patients présentant une surcharge pondérale et traités à la metformine après qu'un régime alimentaire se fut avéré insuffisant à lui seul – a révélé:
- Dans le groupe sous metformine, une réduction significative du risque absolu de subir une complication en rapport avec le diabète (29,8 événements sur 1000 patients-années) par rapport au régime alimentaire seul (43,3 événements sur 1000 patient-années), p = 0,0023, ainsi que par rapport aux groupes combinés sous monothérapie aux sulfonylurées ou à l'insuline (40,1 événements sur 1000 patient-année), p= 0,0034.
- Une réduction significative du risque absolu de mortalité en rapport avec le diabète: metformine 7,5 décès sur 1000 patient-années, régime alimentaire seul 12,7 décès sur 1000 patient-années, p= 0,017.
- Une réduction significative du risque absolu de mortalité totale: metformine 13,5 décès sur 1000 patient-années par rapport au régime alimentaire seul 20,6 sur 1000 patient-années (p= 0,011) et par rapport aux groupes combinés de monothérapie aux sulfonylurées ou à l'insuline 18,9 sur 1000 patient-années (p= 0,021).
- Une réduction significative du risque absolu de subir un infarctus du myocarde: metformine 11 événements sur 1000 patient-années, régime alimentaire seul 18 décès sur 1000 patient-années (p= 0,01).
Étude de sécurité cardiovasculaire TECOS
L'étude TECOS (Trial Evaluating Cardiovascular Outcomes with Sitagliptin) était randomisée et incluait 14'671 patients de la population en intention de traiter présentant des valeurs HbA1c de ≥6,5 à 8,0% et une maladie cardiovasculaire documentée. Les patients ont reçu soit 100 mg par jour de sitagliptine (7332) (ou 50 mg par jour si le DFGe initial était ≥30 et <50 ml/min/1,73 m2) soit un placebo (7339), donné en complément du traitement conventionnel avec des valeurs cibles correspondant aux standards régionaux en termes d'HbA1c et de facteurs de risque cardiovasculaire. Les patients présentant un DFGe <30 ml/min/1,73 m2 n'ont pas été inclus dans l'étude. L'étude incluait 2004 patients âgés de ≥75 ans et 3324 patients insuffisants rénaux (DFGe <60 ml/min/1,73 m2).
Les patients du groupe sitagliptine ont reçu moins d'antihyperglycémiants que ceux du groupe placebo (Hazard Ratio 0,72; IC à 95%, 0,68-0,77; p ≤0,001). Par ailleurs, les patients ne recevant pas d'insuline au début de l'étude étaient moins susceptibles de démarrer une insulinothérapie chronique (Hazard Ratio 0,70; IC à 95%, 0,63-0,79; p <0,001).
Le critère d'évaluation cardiovasculaire primaire était composé de la première survenue d'un décès d'origine cardiovasculaire, d'un infarctus du myocarde non fatal, d'un accident vasculaire cérébral (AVC) non fatal ou d'une hospitalisation en raison d'une angine de poitrine instable. Les critères d'évaluation cardiovasculaires secondaires étaient la première survenue d'un décès d'origine cardiovasculaire, d'un infarctus du myocarde non fatal ou d'un AVC non fatal; la première survenue d'un des composants du critère d'évaluation primaire; la mortalité totale; et les hospitalisations en raison d'une insuffisance cardiaque.
Après une période d'observation médiane de trois ans, le risque d'événements cardiovasculaires indésirables sévères ou d'hospitalisation en raison d'une insuffisance cardiaque n'a pas augmenté lorsque la sitagliptine était administrée chez les patients diabétiques de type 2 en complément du traitement conventionnel (tableau 6).
Tableau 6:
Taux d'événements cardiovasculaires et événements secondaires importants
Sitagliptine 100 mg | Placebo | Hazard Ratio (IC à 95%) | Valeur p† | |||
N (%) | Taux d'incidence pour 100 patients-années* | N (%) | Taux d'incidence pour 100 patients-années | |||
Analyse de la population en intention de traiter | ||||||
Nombre de patients | 7332 | 7339 | 0,98 (0,89–1,08) | <0,001 | ||
Critère d'évaluation primaire composite (décès cardiovasculaire, infarctus du myocarde non fatal, AVC non fatal ou hospitalisation en raison d'une angine de poitrine instable) | 839 (11,4) | 4,1 | 851 (11,6) | 4,2 | ||
Critère d'évaluation secondaire composite (décès cardiovasculaire, infarctus du myocarde non fatal ou AVC non fatal) | 745 (10,2) | 3,6 | 746 (10,2) | 3,6 | 0,99 (0,89–1,10) | <0,001 |
Événements secondaires | ||||||
Décès cardiovasculaires | 380 (5,2) | 1,7 | 366 (5,0) | 1,7 | 1,03 (0,89-1,19) | 0,711 |
Tous les infarctus du myocarde (fatals et non fatals) | 300 (4,1) | 1,4 | 316 (4,3) | 1,5 | 0,95 (0,81–1,11) | 0,487 |
Tous les AVC (fatals et non fatals) | 178 (2,4) | 0,8 | 183 (2,5) | 0,9 | 0,97 (0,79–1,19) | 0,760 |
Hospitalisations en raison d'une angine de poitrine instable | 116 (1,6) | 0,5 | 129 (1,8) | 0,6 | 0,90 (0,70–1,16) | 0,419 |
Décès toutes causes | 547 (7,5) | 2,5 | 537 (7,3) | 2,5 | 1,01 (0,90–1,14) | 0,875 |
Hospitalisations en raison d'une insuffisance cardiaque‡ | 228 (3,1) | 1,1 | 229 (3,1) | 1,1 | 1,00 (0,83–1,20) | 0,983 |
* Le taux d'incidence pour 100 patients-années a été calculé selon la formule 100× (nombre total de patients présentant ≥1 événement pendant la période d'exposition décisive sur le nombre total de patients-années de l'observation).
† D'après le modèle de Cox stratifié par région. Pour les critères d'évaluation composites, les valeurs p correspondent à un test de non-infériorité dont l'objectif est de montrer que le Hazard Ratio est inférieur à 1,3. Pour tous les autres critères d'évaluation, les valeurs p correspondent à un test de différence des Hazard Rates.
‡ L'analyse des hospitalisations en raison d'une insuffisance cardiaque a été ajustée pour l'antécédent d'insuffisance cardiaque à l'inclusion dans l'étude.
Pharmacocinétique
Les résultats d'une étude auprès de volontaires sains ont montré que les comprimés associés Janumet XR (phosphate de sitagliptine/chlorhydrate de metformine à libération retardée) 50 mg/500 mg et 100 mg/1000 mg sont bioéquivalents à une prise simultanée des doses correspondantes de phosphate de sitagliptine (Januvia) et chlorhydrate de metformine à libération retardée sous forme de comprimés séparés.
De même, la bioéquivalence entre deux comprimés de Janumet XR 50 mg/500 mg et un comprimé de Janumet XR 100 mg/1000 mg a été démontrée.
Dans une étude croisée auprès de volontaires sains, l'AUC et la Cmax de la sitagliptine et l'AUC de la metformine étaient similaires après l'administration d'un seul comprimé Janumet XR de 50 mg/500 mg (formule testée) et après l'administration d'un seul comprimé Janumet (phosphate de sitagliptine/chlorhydrate de metformine à libération non retardée) de 50 mg/500 mg. Après administration d'un seul comprimé Janumet XR 50 mg/500 mg (formule testée), la valeur moyenne de Cmax de la metformine était inférieure de 30% et la valeur médiane de Tmax était retardée de 4 heures par rapport aux valeurs correspondantes observées après l'administration d'un seul comprimé Janumet 50 mg/500 mg. Ceci est en accord avec les caractéristiques attendues de la libération retardée de la metformine de Janumet XR.
Après administration de deux comprimés Janumet XR 50 mg/1000 mg une fois par jour avec le repas du soir pendant 7 jours chez des volontaires sains adultes, la sitagliptine et la metformine ont atteint l'état d'équilibre le jour 4 et le jour 5, respectivement. Les valeurs médianes de Tmax pour la sitagliptine et la metformine à l'état d'équilibre étaient d'environ 3 et 8 heures, respectivement, après la prise. Les valeurs médianes de Tmax pour la sitagliptine et la metformine après administration d'un seul comprimé Janumet étaient de 3 et de 3,5 heures, respectivement, après la prise.
Les données pharmacocinétiques caractérisant la distribution, le métabolisme et l'élimination de la metformine et de la sitagliptine proviennent d'études avec administration des monosubstances de metformine à libération non retardée et de sitagliptine; elles sont applicables à Janumet XR.
Absorption
Janumet XR
Après administration de comprimés Janumet XR avec un petit-déjeuner riche en graisses, l'AUC de la sitagliptine était inchangée. La Cmax moyenne était diminuée de 17%, tandis que la valeur médiane de Tmax était inchangée par rapport à l'état à jeun. Après administration de Janumet XR avec un petit-déjeuner riche en graisses, l'AUC de la metformine était augmentée de 62%, la Cmax de la metformine était diminuée de 9% et la valeur médiane de Tmax de la metformine était retardée de 2 heures par rapport à l'état à jeun.
Phosphate de sitagliptine
La biodisponibilité absolue de la sitagliptine se monte à environ 87%. La prise simultanée d'un repas riche en graisses n'a aucune influence sur la pharmacocinétique de la sitagliptine.
Distribution
Phosphate de sitagliptine
Le volume de distribution moyen à l'état stationnaire après une dose intraveineuse unique de 100 mg de sitagliptine chez des volontaires sains se monte à environ 198 litres. La fraction de sitagliptine liée de manière réversible aux protéines plasmatiques est faible (38%).
Chlorhydrate de metformine
Aucune étude n'a été effectuée sur la distribution de la metformine à libération retardée. Le volume de distribution apparent (V/F) de la metformine après la prise de doses orales uniques de 850 mg de chlorhydrate de metformine à libération non retardée a été en moyenne de 654 ± 358 l. Contrairement aux sulfonylurées, qui se lient à plus de 90% aux protéines plasmatiques, la metformine ne présente guère de liaison aux protéines plasmatiques. La metformine se répartit dans les érythrocytes, probablement en fonction du temps. Aux schémas posologiques et dosages cliniques normaux des comprimés de chlorhydrate de metformine, les concentrations plasmatiques de metformine à l'état stationnaire – normalement inférieures à 1 µg/ml – sont atteintes en l'espace de 24 à 48 heures. Dans des études cliniques contrôlées sur la metformine, les taux maximaux de metformine n'ont jamais dépassé 5 µg/ml même aux doses maximales.
Métabolisme
Phosphate de sitagliptine
La sitagliptine est éliminée en première ligne sous forme inchangée dans l'urine et seule une faible fraction est métabolisée. Environ 79% de la sitagliptine sont éliminés dans l'urine sous forme inchangée.
Après une dose orale de [14C]sitagliptine, près de 16% de la radioactivité a été éliminée sous forme de métabolites de la sitagliptine. Six métabolites ont été détectés sous forme de traces. On ne pense pas que ces métabolites ont contribué à l'activité inhibitrice plasmatique de la DPP-4 par la sitagliptine. Des études in vitro ont pu montrer que le CYP3A4, avec la participation du CYP2C8, est la principale enzyme pour le métabolisme limité de la sitagliptine.
Chlorhydrate de metformine
Des études avec des doses intraveineuses uniques chez des volontaires sains montrent que la metformine est éliminée dans l'urine sous forme inchangée, sans être métabolisée dans le foie (aucun métabolite n'a été trouvé chez l'homme) ni éliminée dans la bile.
Élimination
Phosphate de sitagliptine
Après l'administration d'une dose orale de [14C]sitagliptine à des volontaires sains, près de 100% de la radioactivité administrée a été retrouvée dans les selles (13%) ou l'urine (87%) en l'espace d'une semaine. La demi-vie terminale apparente t½ après une dose orale de 100 mg de sitagliptine était de près de 12,4 heures, et la clairance rénale se montait à près de 350 ml/min.
L'élimination de la sitagliptine se fait principalement par les reins et implique une sécrétion tubulaire active. La sitagliptine est un substrat pour le transporteur d'anions organiques 3 de l'homme (hOAT-3), qui est probablement impliqué dans l'élimination rénale de la sitagliptine. L'importance clinique du hOAT-3 pour le transport de la sitagliptine n'a pas encore pu être démontrée. La sitagliptine est également un substrat de la p-glycoprotéine, qui pourrait aussi être impliquée dans le mécanisme de l'élimination rénale de la sitagliptine. La ciclosporine, un inhibiteur de la glycoprotéine p, n'a toutefois pas permis de diminuer la clairance rénale de la sitagliptine.
Chlorhydrate de metformine
La clairance rénale est environ 3,5 fois supérieure à la clairance de la créatinine, ce qui permet de supposer que la sécrétion tubulaire est la voie d'élimination la plus importante de la metformine. Après une administration orale de metformine à libération non retardée, environ 90% de la dose assimilée sont éliminés dans les premières 24 h par voie rénale, avec une demi-vie d'élimination plasmatique de 6,2 heures environ. Dans le sang, la demi-vie d'élimination est d'environ 17,6 heures, ce qui permet de supposer que la masse érythrocytaire pourrait être un compartiment de distribution.
Trouble de la fonction rénale
Phosphate de sitagliptine
Chez les patients présentant un trouble modéré de la fonction rénale, avec un DFGe de 30 à <45 ml/min/1.73 m2, une multiplication par deux environ de l'AUC plasmatique de la sitagliptine a été observée, et chez les patients présentant un trouble sévère de la fonction rénale (DFGe <30 ml/min/1.73 m2), y compris les patients atteints d'insuffisance rénale terminale (end-stage renal disease, ESRD) sous hémodialyse, une augmentation d'environ quatre fois a été observée par rapport aux volontaires dont la fonction rénale était normale.
Chlorhydrate de metformine
Chez les patients présentant une diminution de la fonction rénale, la demi-vie plasmatique et sanguine de la metformine à libération non retardée est prolongée et la clairance rénale est diminuée (voir «Contre-indications» et «Mises en garde et précautions»).
Trouble de la fonction hépatique
Phosphate de sitagliptine
Chez les patients présentant un trouble modéré de la fonction hépatique (Child-Pugh Score 7 à 9), les valeurs moyennes de l'AUC et de la Cmax de la sitagliptine avaient augmenté de 21% et de 13% par rapport au groupe de contrôle après la prise d'une dose unique de 100 mg de sitagliptine. Ces effets ne sont pas considérés comme cliniquement significatifs.
Il n'existe aucune donnée clinique relative aux patients présentant un trouble sévère de la fonction hépatique (Child-Pugh Score >9). Toutefois, étant donné que la sitagliptine est surtout éliminée par les reins, un trouble sévère de la fonction hépatique n'influence probablement pas la pharmacocinétique de la sitagliptine.
Chlorhydrate de metformine
Il n'existe aucune étude pharmacocinétique sur la metformine chez les patients présentant un trouble de la fonction hépatique.
Utilisation chez les personnes âgées
Phosphate de sitagliptine
Selon les données de phase I et II d'une analyse pharmacocinétique de population, l'âge n'a pas d'effet cliniquement significatif sur la pharmacocinétique de la sitagliptine. Les personnes âgées (de 65 ans à 80 ans) ont présenté, par rapport à des personnes plus jeunes, des concentrations plasmatiques de sitagliptine supérieures de près de 19%.
Chlorhydrate de metformine
Il n'existe que peu de données d'études pharmacocinétiques contrôlées sur la metformine à libération non retardée chez les sujets âgés sains. Elles suggèrent que chez les personnes âgées saines, la clairance plasmatique totale de la metformine est réduite, la demi-vie prolongée et la Cmax accrue en comparaison avec les sujets sains encore jeunes.
Ces données font supposer que l'influence de l'âge des patients sur la pharmacocinétique de la metformine provient essentiellement d'altérations de la fonction rénale (voir l'information professionnelle de GLUCOPHAGE1).
1 GLUCOPHAGE® est une marque déposée de Merck Sante S.A.S, société partenaire de Merck KGaA à Darmstadt, Allemagne.
Emploi chez les enfants et les adolescents
Aucune étude n'a examiné la sécurité et l'efficacité de Janumet XR chez les enfants et les adolescents.
Sexe
Phosphate de sitagliptine
Une analyse pharmacocinétique de données de phase I et une analyse de pharmacocinétique de population basée sur des données de phase I et II ont montré que le sexe n'a pas d'effet cliniquement significatif sur la pharmacocinétique de la sitagliptine.
Chlorhydrate de metformine
Les paramètres pharmacocinétiques de la metformine n'ont pas été notablement différents entre les sujets sains et les patients diabétiques de type 2, lorsqu'ils ont été analysés en fonction du sexe des patients. De même, des études cliniques contrôlées auprès de patients diabétiques de type 2 ont permis de montrer que les effets anti-hyperglycémiants de la metformine sont similaires chez l'homme et la femme.
Race
Phosphate de sitagliptine
Une analyse pharmacocinétique basée sur des données de phase I et une analyse pharmacocinétique de population basée sur des données de phase I et II, ayant inclus des Caucasiens, des Latino-américains, des Afro-américains, des Asiatiques et des membres d'autres groupes ethniques ont montré que l'appartenance ethnique n'a pas d'effet cliniquement significatif sur la pharmacocinétique de la sitagliptine.
Chlorhydrate de metformine
Il n'existe aucune étude sur les paramètres pharmacocinétiques de la metformine en fonction de la race. Dans des études cliniques contrôlées sur la metformine chez des patients diabétiques de type 2, les effets anti-hyperglycémiants ont été comparables entre les Caucasiens (n= 249), les Afro-américains (n= 51) et les Latino-américains (n= 24).
Indice de masse corporelle (Body-Mass-Index, BMI)
Phosphate de sitagliptine
Une analyse pharmacocinétique de données de phase I et une analyse de pharmacocinétique de population sur la base de données de phase I et II ont montré que le BMI n'a pas d'effet cliniquement significatif sur la pharmacocinétique de la sitagliptine.
Données précliniques
Sitagliptine et metformine
Des études précliniques toxicocinétiques et de toxicité orale ont été effectuées chez le chien avec la sitagliptine et la metformine sous forme d'association fixe à libération non retardée.
Dans une étude de toxicité orale de 16 semaines, des chiennes ont été traitées à la metformine 20 mg/kg/jour seule ou associée à 2, 10 ou 50 mg/kg/jour de sitagliptine. Une ataxie transitoire et/ou un tremblement ont été observés dans le groupe sous forte dose. Ces signes ont été jugés imputables à la sitagliptine, étant donné qu'ils avaient été observés dans d'autres études sur des chiens sous sitagliptine seule dosée à 50 mg/kg/jour. Dans cette étude, le niveau d'effet nul (no-effect level) pour les effets imputables au traitement a été de 10 mg/kg/jour de sitagliptine et de 20 mg/kg/jour de metformine, ce qui correspond chez l'homme à 6 fois l'exposition systémique à une dose journalière de 100 mg de sitagliptine et à 2,5 fois l'exposition systémique à une dose journalière de 2000 mg de metformine.
Aucune étude préclinique n'a été effectuée avec le médicament associé Janumet XR sur le potentiel cancérigène, la mutagénicité, la perturbation de la fertilité ou les effets sur la reproduction. Les données suivantes proviennent d'études séparées sur la sitagliptine seule et la metformine seule.
Sitagliptine
Lors d'études non cliniques chez des rats, des effets toxiques de la sitagliptine n'ont été observés qu'à des expositions largement supérieures à la dose humaine, de sorte qu'ils ne doivent pas être pris en compte pour l'utilisation normale chez l'être humain. Au cours d'études précliniques sur la sécurité, la sitagliptine ne s'est avérée ni génotoxique, ni mutagène.
Dans une série d'études sur la toxicité avec des administrations répétées du médicament à des chiens, des doses de 2, 10 et 50 mg/kg/jour ont été examinées pendant une période pouvant aller jusqu'à 53 semaines. Après 53 semaines de traitement avec une dose de 10 mg/kg/jour, ce qui correspond à six fois la dose recommandée pour l'adulte, de 100 mg/jour, aucun effet n'a été constaté. Chez des chiens ayant reçu la dose de 50 mg/kg/jour de sitagliptine (environ 26 fois l'exposition humaine), des symptômes physiques associés au traitement sont apparus passagèrement, par exemple une respiration avec la bouche ouverte, une salivation, des vomissements blanchâtres et mousseux, une ataxie, des tremblements, une diminution des activités et/ou une position du corps courbée. Lors des études sur la toxicité, de 14 et 27 semaines, avec une dose de 50 mg/kg/jour, une dégénérescence très légère à légère de la musculature squelettique a pu en outre être observée au niveau histologique. Au cours des études sur la toxicité de 53 semaines toutefois, aucune dégénérescence de la musculature squelettique n'a pu être constatée, ce qui laisse supposer qu'il s'agit d'un effet non reproductible ou que la modification n'apparaît plus avec un traitement prolongé. L'exposition systémique dans le domaine NOEL était lors de l'étude à 53 semaines (10 mg/kg/jour) 5 fois plus élevée que dans l'étude à 27 semaines (2 mg/kg/jour).
D'autres études de toxicité orale sur 3 mois ont été effectuées chez des singes rhésus et cynomolgus. L'étude de 3 mois chez le singe rhésus a examiné le potentiel de lésions cutanées ou de toxicité rénale de la sitagliptine, l'évaluation s'est limitée à la peau et aux reins. Les animaux ont reçu jusqu'à 100 mg de sitagliptine/kg/jour (28 fois l'exposition systémique d'une dose quotidienne de 100 mg). Il n'y a pas eu d'observations ante mortem ou post mortem dues au traitement.
Dans l'étude de toxicité de 3 mois menée chez le singe cynomolgus, les évaluations de routine complètes ont été effectuées. Les animaux ont reçu jusqu'à 100 mg de sitagliptine/kg/jour (27 fois l'exposition systémique d'une dose quotidienne de 100 mg). Il n'y a pas eu d'observations ante mortem ou post mortem dues au traitement.
La sitagliptine s'est avérée non cancérigène chez des souris ayant reçu pendant deux ans la dose maximale tolérée, de 500 mg/kg/jour, par voie orale. Une étude sur la cancérogénicité, de deux ans, a été réalisée avec des rats du sexe masculin et féminin, qui ont reçu des doses orales de 50, 150 et 500 mg/kg/jour de sitagliptine. Une augmentation des cas d'adénomes hépatiques et de cancers a été observée chez les rats du sexe masculin et une hausse des cas de cancers du foie a été constatée chez des rats du sexe féminin ayant reçu des doses élevées. Sur la base de la dose journalière recommandée chez l'être humain, de 100 mg/jour, cette dose correspondait à 58 fois l'exposition humaine chez des rats. Cette dose était associée, lors d'essais chez les rats, à une hépatotoxicité. Le No-observed-Effect-Level pour l'induction d'une néoplasie hépatique était de 150 mg/kg/jour, ce qui correspond à 19 fois l'exposition humaine à la dose recommandée de 100 mg. Étant donné qu'il a pu être montré que, chez le rat, l'induction d'une néoplasie hépatique était en corrélation avec l'hépatotoxicité, on peut considérer que la fréquence plus élevée des tumeurs hépatiques chez les rats était une conséquence de la toxicité hépatique chronique sous cette dose élevée. La signification clinique de ce résultat pour l'homme n'est pas connue.
Aucun effet négatif sur la fertilité de rats du sexe masculin et féminin ayant reçu de la sitagliptine par voie orale avant ou durant l'accouplement, à des doses pouvant aller jusqu'à 1000 mg/kg/jour (correspondant à près de cent fois l'exposition chez l'être humain, sur la base de la dose journalière recommandée, de 100 mg/jour pour l'adulte) n'a été observé.
Des études de toxicologie de la reproduction réalisées chez des rats ayant reçu des doses orales de 1000 mg/kg/jour ont montré une légère augmentation associée au traitement de la fréquence de malformations fœtales des côtes (côtes manquantes, hypoplasie et côtes déformées) dans la descendance. Le No-observed-Effect-Level pour les troubles du développement était de 250 mg/kg/jour (ce qui correspond à 32 fois l'exposition chez l'être humain, sur la base de la dose journalière recommandée, de 100 mg/jour chez l'adulte). La sitagliptine est excrétée dans le lait de rates allaitantes. Aucune étude n'a été effectuée avec la sitagliptine, la metformine et une sulfonylurée.
Chlorhydrate de metformine
Les données précliniques d'études conventionnelles de pharmacologie de sécurité, d'études de toxicité avec administrations répétées du médicament et d'études sur la génotoxicité, le potentiel carcinogène et la toxicité sur la reproduction montrent qu'aucun danger spécifique ne découle d'une prise de la metformine chez l'homme.
Remarques particulières
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Le médicament doit être conservé hors de la portée des enfants.
Ne pas conserver au-dessus de 30 °C. Conserver le flacon hermétiquement fermé à l'abri de l'humidité.
Numéro d’autorisation
65052 (Swissmedic).
Titulaire de l’autorisation
MSD MERCK SHARP & DOHME AG, Lucerne.
Mise à jour de l’information
Septembre 2020
HMV4+rhabdomyolysis / RCN000010607-CH+RCN000019721-CH
Reviews (0)

Free consultation with an experienced pharmacist
Describe the symptoms or the right drug - we will help you choose its dosage or analogue, place an order with home delivery or just consult.
We are 14 pharmacists and 0 bots. We will always be in touch with you and will be able to communicate at any time.
Bestsellers

Milupa Aptamil Pepti Syneo Can 400g
Product code: 7748334Aptamil Pepti Syneo is a special food for babies from birth who are allergic to cow's milk protein a..
55.08 CHF


Weleda Baby Calendula Wind- und Wetterbalsam 30ml
Product code: 5455461Der Weleda Baby Calendula Wind- und Wetterbalsam schützt die sensible Babyhaut intensiv bei Kälte un..
15.35 CHF


HerpoTherm herpes pen
Product code: 7798882Herpotherm® - the heating pen Cold sores are not only unattractive but can also be very painful. Th..
78.48 CHF



