




Imnovid Kaps 2 mg 21 pcs
Imnovid Kaps 2 mg 21 Stk
-
12,499.88 CHF
- Price in reward points: 3131

- Availability: Not available
- Brand: CELGENE GMBH
- Product Code: 6011068
- ATC-code L04AX06
- EAN 7680612490029
Ingredients:
Erythrosin (E127), Gelatine, Mannitol, Propylenglycol, Titandioxid (E171), Indigocarmin (E132), Natrium 0.036 mg, Simeticon, Eisen(III)-oxid (E172), Ammoniaklösung 10 %, Natriumstearylfumarat, Schellack, Drucktinte, Stärke vorverkleistert, Pomalidomid 2 mg , Kapselhülle.

Description
 Deutsch
Deutsch French
French Italian
ItalianWas ist Imnovid und wann wird es angewendet?
Auf Verschreibung des Arztes oder der Ärztin.
Imnovid enthält den Wirkstoff Pomalidomid. Dieses Arzneimittel gehört zu einer Gruppe von Arzneimitteln, die die Funktionsweise des körpereigenen Immunsystems beeinflussen.
Imnovid wird bei Patienten mit multiplem Myelom (Tumorerkrankung mit starker Vermehrung von gewissen Zellen im Knochenmark) folgendermassen eingesetzt:
- entweder zusammen mit Bortezomib (ein sogenannter Proteasom-Inhibitor) und Dexamethason (ein entzündungshemmendes Arzneimittel) bei Patienten, welche bereits zuvor eine oder mehr Behandlungen erhalten haben,
- oder nur zusammen mit Dexamethason bei Patienten, welche bereits zuvor zwei oder mehr Behandlungen erhalten haben.
Wann darf Imnovid nicht eingenommen werden?
Falls Sie schwanger sind oder denken, Sie könnten schwanger sein, oder eine Schwangerschaft planen.
Bei gebärfähigen Frauen, es sei denn, es werden strenge Massnahmen zur Schwangerschaftsverhütung ergriffen; siehe «Wann ist bei der Einnahme von Imnovid Vorsicht geboten?».
Bei Überempfindlichkeit gegenüber dem Wirkstoff Pomalidomid oder einem der Hilfsstoffe oder gegenüber Thalidomid oder Lenalidomid.
Wann ist bei der Einnahme von Imnovid Vorsicht geboten?
Für Frauen, welche Imnovid einnehmen
Schwangerschaftsverhütung
Der Arzt bzw. die Ärztin kann Frauen, welche schwanger werden können, in besonderen Fällen Imnovid verschreiben. Falls dies bei Ihnen der Fall ist, ist es unbedingt notwendig, dass Sie die Anordnungen Ihres Arztes bzw. Ihrer Ärztin exakt befolgen. Die folgenden Punkte sind sehr wichtig:
- Vor Beginn der Behandlung wird Ihr Arzt bzw. Ihre Ärztin überprüfen, dass Sie nicht schwanger sind und er/sie wird ausserdem während der gesamten Behandlungszeit inklusive zeitweiligen Behandlungsunterbrüchen alle 4 Wochen sowie 4 Wochen nach Ende der Behandlung einen Schwangerschaftstest durchführen.
- Ihr Arzt bzw. Ihre Ärztin oder ein anderer Arzt bzw. eine andere Ärztin wird Sie über geeignete Methoden zur Schwangerschaftsverhütung beraten. Sie müssen mit diesen Methoden im Allgemeinen 4 Wochen vor Behandlungsbeginn anfangen und diese nicht nur während der Behandlungsdauer inkl. zeitweisen Behandlungsunterbrüchen, sondern auch während vier Wochen nach Ende der Behandlung strikt einhalten.
- Falls Sie trotz strikter Einhaltung der Massnahmen zur Schwangerschaftsverhütung während der Behandlung mit Imnovid oder innerhalb eines Monats nach Abschluss der Behandlung schwanger werden, oder vermuten, Sie könnten schwanger sein, müssen Sie Ihren Arzt bzw. Ihre Ärztin sofort benachrichtigen. Ihr Arzt bzw. Ihre Ärztin wird dann die notwendigen Massnahmen einleiten.
Für Männer, welche Imnovid einnehmen
Männliche Patienten, welche mit einer Frau im gebärfähigen Alter Geschlechtsverkehr haben, müssen während der Behandlungsdauer inkl. zeitweisen Unterbrüchen und während 7 Tagen nach Ende der Behandlung Kondome verwenden. Ferner dürfen Sie während der Behandlung mit Imnovid und 7 Tage danach kein Sperma spenden.
Wegen des Risikos für das ungeborene Kind dürfen Sie Imnovid niemals an andere Personen weitergeben.
Die Zulassungsinhaberin von Imnovid stellt folgendes Material zur Verfügung:
- Information über die Schwangerschaftsproblematik
- Patientenbroschüre mit einem Formular, welches Sie unterschreiben müssen, um zu bestätigen, dass Sie die Notwendigkeit, eine Schwangerschaft unter Therapie mit Imnovid zu verhindern, verstanden haben.
Andere Vorsichtsmassnahmen
Ihr Arzt bzw. Ihre Ärztin wird vor und während der Behandlung mit Imnovid bei Ihnen regelmässig Blutuntersuchungen durchführen, da Ihr Arzneimittel dazu führen kann, dass die Zahl der Blutkörperchen, die helfen, eine Infektion zu bekämpfen, und die für die Blutgerinnung verantwortlich sind, abnehmen können.
In Abhängigkeit von den Ergebnissen Ihrer Blutuntersuchungen und Ihres Allgemeinzustandes wird Ihr Arzt eventuell die Imnovid-Dosis anpassen oder die Behandlung beenden.
Unter der Behandlung mit Imnovid ist das Risiko für die Bildung von Blutgerinnseln in den Gefässen (venöse Thromboembolien) erhöht. Sie sollten sich unverzüglich an Ihren Arzt bzw. Ihre Ärztin wenden, wenn folgende Symptome auftreten sollten: Fieber, Schüttelfrost, Halsweh, Husten, Mundgeschwüre oder andere Infektionssymptome, Blutungen oder blaue Flecken ohne erkennbare Ursache, Schmerzen an einem Bein oder Brustschmerzen, Kurzatmigkeit.
Bei Patienten mit vorbestehender Herzerkrankung oder kardialen Risikofaktoren kann es zu Herzschwäche kommen. Wenden Sie sich an Ihren Arzt bzw. Ihre Ärztin, falls Sie bereits einen Herzinfarkt hatten, an einer Herzmuskelschwäche oder Atemschwierigkeiten leiden oder wenn Sie rauchen, einen hohen Blutdruck oder hohen Cholesterinspiegel aufweisen.
Vor allem bei Patienten, die an einer grösseren Tumorlast vor Behandlungsbeginn mit Imnovid leiden, kann als Folge des schnellen Zerfalls der Krebszellen ein sogenanntes Tumorlyse-Syndrom auftreten. Wenden Sie sich an Ihren Arzt bzw. Ihre Ärztin falls Sie Symptome wie Übelkeit, Atemnot, unregelmässiger Herzschlag, eingetrübter Urin, Müdigkeit und/oder Gelenkbeschwerden bemerken.
Mögliche schwerwiegende allergische Reaktionen (genannt Angioödem und Anaphylaxie) können sich in Form von Nesselsucht, Hautausschlag, Anschwellen von Augen, Mund oder Gesicht, Atemnot oder Juckreiz manifestieren. Schwerwiegende allergische Reaktionen können zu Beginn als örtlich begrenzte Hautausschläge auftreten, die sich dann über den gesamten Körper ausbreiten verbunden mit grossflächigen Hautablösungen (genannt Steven-Johnson Syndrom und/oder toxische epidermale Nekrolyse). In sehr seltenen Fällen können allergische Reaktionen zusätzlich zu Hautreaktionen von Fieber, Müdigkeit, Schwellung der Lymphknoten, Anstieg bestimmter weisser Blutkörperchen (Eosinophilie) sowie Auswirkungen auf Leber, Niere oder Lunge begleitet sein (genannt DRESS).
Imnovid sollte nicht gleichzeitig mit starken CYP1A2-Hemmern (z.B. Ciprofloxacin) eingenommen werden. Informieren Sie Ihren Arzt bzw. Ihre Ärztin wenn Sie starke CYP1A2-Hemmer (z.B. Ciprofloxacin) einnehmen. Diese Medikamente beeinflussen den Abbau von Imnovid im Körper. Aus diesem Grund wird Ihr Arzt bzw. Ihre Ärztin Ihre Imnovid-Dosis wenn nötig anpassen.
Bei Patienten unter Behandlung mit Imnovid sind erhöhte Leberfunktionswerte im Blut beobachtet worden. Auch gab es Fälle von Leberentzündungen, welche zu einem Behandlungsabbruch geführt haben. Aus diesem Grund wird Ihre Leberfunktion regelmässig durch Ihren Arzt bzw. Ihre Ärztin gemessen.
Auf Grund von möglichen Nebenwirkungen wie Müdigkeit, Schwindel und Verwirrtheit, sollten Sie beim Lenken eines Fahrzeugs oder beim Bedienen von Maschinen besonders vorsichtig sein.
Es ist wichtig zu beachten, dass es bei einer geringen Anzahl von Patienten mit multiplem Myelom zur Entwicklung weiterer Krebsarten kommen kann, und es ist möglich, dass sich dieses Risiko bei einer Behandlung mit Imnovid erhöht. Daher wird Ihr Arzt bzw. Ihre Ärztin vor der Verordnung von Imnovid eine sorgfältige Nutzen-Risiko-Bewertung vornehmen.
Bei einer geringen Anzahl von Patienten, die zuvor mit dem Hepatitis-B-Virus infiziert worden waren, wurde bei einer Behandlung mit Imnovid in Kombination mit Dexamethason eine Reaktivierung von Hepatitis B beobachtet. Daher wird Sie Ihr Arzt bzw. Ihre Ärztin während Ihrer Therapie mit Imnovid sorgfältig auf Anzeichen und Symptome einer aktiven Hepatitis-B-Vireninfektion untersuchen. Informieren Sie Ihren Arzt bzw. Ihre Ärztin wenn Sie in der Vergangenheit eine Hepatitis-B-Vireninfektion hatten.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Kapsel, d.h. es ist nahezu «natriumfrei».
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wenn Sie an anderen Krankheiten leiden, Allergien haben oder andere Arzneimittel (auch selbst gekaufte!) einnehmen oder äusserlich anwenden.
Darf Imnovid während einer Schwangerschaft oder in der Stillzeit eingenommen werden?
Imnovid darf während einer Schwangerschaft nicht eingenommen werden.
Gebärfähige Frauen sollen während einer Behandlung mit Imnovid nicht schwanger werden. Zum Ausschluss einer Schwangerschaft müssen vor, während und bei Ende der Behandlung Schwangerschaftstests durchgeführt werden und es sind wirksame Methoden zur Empfängnisverhütung anzuwenden.
Es ist nicht bekannt, ob Imnovid in die Muttermilch übertritt. Daher soll Imnovid während der Stillzeit nicht angewendet werden oder es soll abgestillt werden.
Pomalidomid tritt in die menschliche Samenflüssigkeit über. Männliche Patienten mit einer gebärfähigen Partnerin müssen während der Behandlung mit Imnovid und während mindestens 7 Tage nach Ende der Behandlung Kondome zur Empfängnisverhütung verwenden.
Detaillierte Angaben betreffend Schwangerschaftsverhütung finden sich unter «Wann ist bei der Einnahme von Imnovid Vorsicht geboten?».
Wie verwenden Sie Imnovid?
Nehmen Sie Imnovid immer genau nach Anweisung des Arztes bzw. der Ärztin ein. Fragen Sie bei Ihrem Arzt oder Apotheker bzw. Ihrer Ärztin oder Apothekerin nach, wenn Sie sich nicht ganz sicher sind.
Nehmen Sie Imnovid jeweils etwa zur selben Tageszeit unabhängig von einer Mahlzeit mit etwas Wasser ein. Die Kapseln sollen nicht zerbrochen, geöffnet oder zerkaut werden. Wenn Pulver aus einer zerbrochenen Pomalidomid-Kapsel mit der Haut in Berührung kommt, reinigen Sie die betroffene Hautstelle sofort gründlich mit Wasser und Seife.
Zur Entnahme der Kapsel aus der Blisterpackung drücken Sie die Kapsel bitte nur an einem Ende aus der Folie heraus. Drücken Sie nicht auf die Mitte der Kapsel, sonst kann die Kapsel zerbrechen.
Imnovid zusammen mit Bortezomib und Dexamethason
Die empfohlene Dosis von Imnovid beträgt 4 mg einmal täglich. Nehmen Sie Imnovid 14 Tage hintereinander ein. Für die folgenden 7 Tage wird die Einnahme von Imnovid unterbrochen. Ein Behandlungszyklus dauert somit 21 Tage. Wiederholen Sie dann dieses Anwendungsschema, bis Ihr Arzt bzw. Ihre Ärztin Sie auffordert, die Einnahme zu beenden.
Ihr Arzt bzw. Ihre Ärztin wird die Dosis von Bortezomib, dem einen Arzneimittel, das Sie in Kombination mit Imnovid verabreicht bekommen, abhängig von Ihrem Gewicht und Ihrer Grösse bestimmen.
Die Dosis von Dexamethason, dem anderen Arzneimittel, das Sie in Kombination mit Imnovid einnehmen müssen, beträgt 20 mg einmal täglich (oder 10 mg einmal täglich, falls Sie über 75 Jahre alt sind), üblicherweise gemäss dem Anwendungsschema wie in der nachfolgenden Tabelle beschrieben:
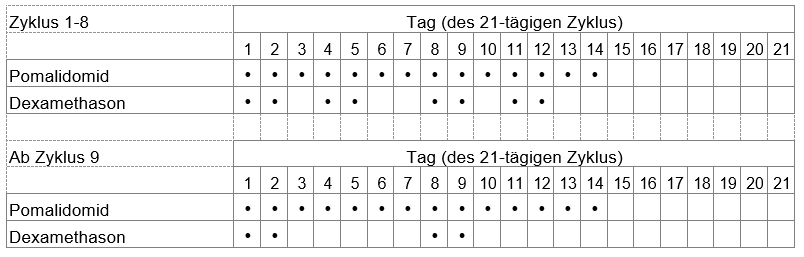
Je nach Beurteilung Ihres Arztes bzw. Ihrer Ärztin kann diese Dosierung individuell angepasst werden.
Weitere Angaben zu Dexamethason finden Sie in der Packungsbeilage des entsprechenden Präparates.
Imnovid zusammen mit Dexamethason
Die empfohlene Dosis von Imnovid beträgt 4 mg einmal täglich. Nehmen Sie Imnovid 21 Tage hintereinander ein. Für die folgenden 7 Tage wird die Einnahme von Imnovid unterbrochen. Ein Behandlungszyklus dauert somit 28 Tage. Wiederholen Sie dann dieses Anwendungsschema, bis Ihr Arzt bzw. Ihre Ärztin Sie auffordert, die Einnahme zu beenden.
Die Dosis von Dexamethason, dem Arzneimittel, das Sie in Kombination mit Imnovid einnehmen müssen, beträgt 40 mg einmal täglich (oder 20 mg einmal täglich, falls Sie über 75 Jahre alt sind). Das Anwendungsschema ist üblicherweise wie folgt: Während den 28-tägigen Behandlungszyklen nehmen Sie Dexamethason an den Tagen 1, 8, 15 und 22 jedes Zyklus ein. Je nach Beurteilung Ihres Arztes bzw. Ihrer Ärztin kann diese Dosierung individuell angepasst werden.
Weitere Angaben zu Dexamethason finden Sie in der Packungsbeilage des entsprechenden Präparates.
Falls Sie eine Dialyse auf Grund einer eingeschränkten Nierenfunktion erhalten, sollten Sie Imnovid an Dialysetagen erst nach der Dialyse einnehmen.
Bei Kindern und Jugendlichen wird Imnovid nicht eingesetzt, da es keine Untersuchungen bei diesen Altersgruppen gibt.
Informieren Sie sofort Ihren Arzt bzw. Ihre Ärztin, wenn Sie eine grössere Menge von Imnovid eingenommen haben, als Ihnen verordnet wurde.
Wenn Sie die Einnahme von Imnovid an einem Tag, an dem Sie das Arzneimittel einnehmen sollen, vergessen haben, nehmen Sie die nächste Kapsel zur gewohnten Zeit am nächsten Tag ein. Erhöhen Sie die Zahl der eingenommenen Kapseln nicht, um die vergessene Dosis Imnovid vom Vortag nachzuholen.
Ändern Sie nicht von sich aus, die verschriebene Dosierung. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. mit Ihrer Ärztin oder Apothekerin.
Welche Nebenwirkungen kann Imnovid haben?
Sehr häufig (betrifft mehr als einen von 10 Anwendern)
Infektionen, einschliesslich Infektionen der Lunge, Nase, Nasennebenhöhlen und des Rachens, Abnahme der Zahl der weissen und roten Blutkörperchen sowie der Blutplättchen (was zu Infektionen, Müdigkeit und Blutungen führen kann), niedriger Kaliumspiegel im Blut, hoher Blutzuckerspiegel, Appetitlosigkeit, Schlafstörungen, Neuropathien (z.B. Schmerzen, Brennen, Taubheits- oder Schwächegefühl, «Ameisenlaufen», Verlust der Tast-, Wärme- oder Schmerzempfindung in den Gliedmassen sowie unkontrollierte oder abnorme Bewegungen), Schwindel, Zittern, Kurzatmigkeit, Husten, Verstopfung, Durchfall, Übelkeit, Erbrechen, Muskelkrämpfe (Spasmen oder Schmerzen), Muskelschwäche, Knochenschmerzen, Rückenschmerzen, Müdigkeitsgefühl, Fieber, Schwächegefühl, Anschwellen der Gliedmassen (Waden, Knöchel), allgemeines Anschwellen wie z.B. des Gesichts.
Häufig (betrifft 1 bis 10 von 100 Anwendern)
Blutvergiftung, Entzündung und Schwellung der Atemwege in der Lunge, Darmentzündung einhergehend mit Unterleibschmerzen und blutigen Durchfällen, Grippe, Harnwegsinfektionen, Pilzinfektionen, Pilzinfektionen im Mund, Basalzellkarzinom, Durstgefühl, niedriger Natriumspiegel im Blut, hoher Kaliumspiegel im Blut, hoher oder niedriger Kalziumspiegel im Blut, niedriger Phosphatspiegel im Blut, niedriger Albumin- oder Magnesiumspiegel im Blut, erhöhter ALT-Spiegel im Blut (ein Leberfunktionswert), erhöhte Kreatininspiegel im Blut (ein Nierenfunktionswert), Verwirrtheit, Angstzustände, Depression, Stimmungsschwankungen, kurz andauernde Bewusstlosigkeit (Synkope), Schläfrigkeit, Benommenheit, Kopfschmerzen, veränderte Geschmackswahrnehmung, verschwommenes Sehen, Trübung der Augenlinse, Veränderungen von Herzfrequenz oder -rhythmus, schneller Herzschlag, hoher oder tiefer Blutdruck, tiefe Venenthrombose (Schmerzen oder Schwellungen in den Beinen, besonders in den Unterschenkeln oder Waden), Lungenembolie (Verstopfung eines Blutgefässes in der Lunge) und andere Lungenprobleme, verstopfte Nase, Rachenschmerzen, Veränderungen der Stimme oder rasselnde Atemgeräusche, Blutungen einschliesslich Nasenbluten, Atemnot unter Belastung, Bauchschmerzen, Entzündung der Mundschleimhaut, trockene Lippen oder trockener Mund, aufgeblähter Bauch, Ausschläge, trockene Haut, juckende Haut, vermehrtes Schwitzen, Schmerzen in den Gelenken oder Extremitäten, Schmerzen im Brustkorb, Bewegungsprobleme, Schmerzen, Nierenversagen, Harnverhalt, Schüttelfrost, Drehschwindel, Beckenschmerzen, Gewichtsverlust, Sturz.
Gelegentlich (betrifft 1 bis 10 von 1000 Anwendern)
Gelbfärbung der Haut und des Augenweisses (Gelbsucht).
Erfahrungen nach Marktzulassung
Verminderung der weissen und roten Blutzellen und der Blutplättchen, Tumorlyse-Syndrom, allergische und schwerwiegende allergische Reaktionen (Angioödem, Anaphylaxie, Steven-Johnson-Syndrom, toxische epidermale Nekrolyse, oder DRESS), erhöhte Leberfunktionswerte und Leberentzündung (z.B. dunkler Urin, Gelbsucht), Reaktivierung des Hepatitis-B-Virus, Herpes Zoster, Gastrointestinale Blutungen, Plattenepithelkarzinom der Haut, Schilddrüsenunterfunktion, Lungenerkrankung einschliesslich Lungenentzündung.
Wenn Sie Nebenwirkungen bemerken, wenden Sie sich an Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Dies gilt insbesondere auch für Nebenwirkungen, die nicht in dieser Packungsbeilage angegeben sind.
Was ist ferner zu beachten?
Das Arzneimittel darf nur bis zu dem auf dem Behälter mit «EXP» bezeichneten Datum verwendet werden.
Lagerungshinweis
Bewahren Sie Imnovid in der Originalverpackung, nicht über 25 °C und ausser Reichweite von Kindern auf.
Weitere Hinweise
Bringen Sie nicht gebrauchte oder beschädigte Kapseln Ihrem Arzt oder Apotheker bzw. Ihrer Ärztin oder Apothekerin zur fachgerechten Entsorgung zurück.
Weitere Auskünfte erteilt Ihnen Ihr Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in Imnovid enthalten?
Wirkstoffe
Pomalidomid: 1 mg, 2 mg, 3 mg oder 4 mg.
Hilfsstoffe
Mannitol, vorverkleisterte Stärke, Natriumstearylfumarat.
Kapselhülle: Gelatine, Titandioxid, gelbes Eisenoxid (nur bei Hartkapseln 1 mg, 2 mg und 3 mg), Indigocarmin E132, Erythrosin E127 (nur bei Hartkapseln 2 mg), Brillantblau FCF E133 (nur bei Hartkapseln 4 mg).
Drucktinte: Shellack, schwarzes Eisenoxid, Titandioxid, Simeticon, Propylenglycol, Ammoniak-Lösung.
Zulassungsnummer
61249 (Swissmedic).
Wo erhalten Sie Imnovid? Welche Packungen sind erhältlich?
In Apotheken gegen ärztliche Verschreibung, die nur zum einmaligen Bezug berechtigt.
Imnovid 1 mg, 2 mg, 3 mg und 4 mg: Packungen zu jeweils 14 oder 21 Hartkapseln.
Zulassungsinhaberin
Celgene GmbH, Zürich.
Diese Packungsbeilage wurde im Januar 2020 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
Qu’est-ce que l’Imnovid et quand doit-il être utilisé?
Selon prescription du médecin.
Imnovid contient la substance active «pomalidomide». Ce médicament appartient à un groupe de médicaments qui modifient le fonctionnement du système immunitaire.
Imnovid est instauré chez des patients atteints d'un myélome multiple (cancer avec une forte prolifération de certaines cellules dans la moelle osseuse) comme suit:
- soit avec le bortézomib (un inhibiteur de protéasome) et la dexaméthasone (un médicament anti-inflammatoire) chez des patients qui ont déjà reçu un ou plusieurs traitements préalables,
- soit uniquement avec la dexaméthasone chez des patients qui ont déjà reçu deux ou plusieurs traitements préalables.
Quand Imnovid ne doit-il pas être pris?
Si vous êtes enceinte, si vous pensez que vous pourriez l'être ou si vous prévoyez de l'être.
Chez les femmes en âge de procréer, sauf si une méthode de contraception très fiable est rigoureusement appliquée; voir la rubrique «Quelles sont les précautions à observer lors de la prise d'Imnovid?».
En cas d'hypersensibilité au pomalidomide, la substance active, ou à l'un des composants ou au thalidomide ou au lénalidomide.
Quelles sont les précautions à observer lors de la prise d’Imnovid?
Pour les femmes qui prennent Imnovid
Mesures contraceptives
Dans des cas particuliers, le médecin peut prescrire Imnovid à des femmes en âge de procréer. Si c'est le cas pour vous, il est absolument nécessaire que vous suiviez exactement les consignes de votre médecin. Les points suivants sont très importants:
- Avant le début du traitement, votre médecin vérifiera que vous n'êtes pas enceinte et en outre, il effectuera un test de grossesse toutes les 4 semaines pendant toute la durée du traitement y compris pendant les interruptions temporaires du traitement et 4 semaines après la fin du traitement.
- Votre médecin ou un autre médecin vous conseillera sur les méthodes contraceptives appropriées. Vous devez utiliser ces méthodes en général 4 semaines avant le début du traitement et continuer à les utiliser à la lettre pendant la durée du traitement y compris les interruptions temporaires du traitement mais aussi jusqu'à 4 semaines après la fin du traitement.
- Si en dépit d'avoir adopté les méthodes contraceptives à la lettre, vous découvrez que vous êtes enceinte ou que vous pensez l'être pendant le traitement par Imnovid ou au cours du mois suivant la fin du traitement, vous devez immédiatement le signaler à votre médecin. Il prendra alors les mesures nécessaires.
Pour les hommes qui prennent Imnovid
Les hommes qui ont des rapports sexuels avec une femme en âge de procréer, doivent utiliser un préservatif pendant la durée du traitement y compris les interruptions temporaires et pendant 7 jours après la fin du traitement. Par ailleurs, ils ne peuvent pas faire de don de sperme pendant le traitement par Imnovid et pendant 7 jours après le traitement.
En raison du risque pour l'enfant à naître, vous ne devez jamais donner Imnovid à d'autres personnes.
Le titulaire de l'autorisation d'Imnovid met à disposition les documents suivants:
- Information sur les problèmes que pose une grossesse
- Brochures pour les patients avec un formulaire que vous devez signer pour confirmer que vous avez compris la nécessité d'éviter une grossesse pendant le traitement par Imnovid.
Autres mesures de précaution
Votre médecin effectuera des analyses de sang à intervalles réguliers avant et pendant le traitement par Imnovid parce que votre médicament peut entraîner une diminution du nombre de cellules sanguines qui contribuent à lutter contre les infections et celles qui sont responsables de la coagulation.
En fonction des résultats de vos analyses de sang et de votre état général, votre médecin pourra éventuellement modifier votre dose d'Imnovid ou arrêter le traitement.
Pendant le traitement par Imnovid, le risque de formation de caillots sanguins dans les vaisseaux sanguins (thrombo-embolies veineuses) est augmenté. Vous devez immédiatement consulter votre médecin si les symptômes suivants se présentent: fièvre, frissons, mal de gorge, toux, ulcères dans la bouche ou d'autres symptômes d'infection, saignements ou taches bleues apparues sans raison, douleur dans une jambe ou douleur dans la poitrine, essoufflement.
Une faiblesse cardiaque peut se manifester chez les patients atteints d'une maladie cardiaque préexistante ou qui ont des facteurs de risque cardiaque. Adressez-vous à votre médecin si vous avez eu une crise cardiaque (infarctus du myocarde), si vous avez une insuffisance cardiaque, si vous avez des difficultés respiratoires, ou si vous fumez, si vous avez une pression artérielle élevée (hypertension) ou un taux de cholestérol élevé.
Suite à la destruction rapide des cellules tumorales, ce qu'on appelle un syndrome de lyse tumorale peut se présenter, en particulier chez les patients qui présentent une charge tumorale élevée avant le début du traitement par Imnovid. Adressez-vous à votre médecin si vous notez des symptômes tels que nausées, essoufflement, battements de cœur irréguliers, urines troubles, fatigue et/ou troubles articulaires.
Les réactions allergiques graves éventuelles (appelées angiœdème et anaphylaxie) peuvent se manifester sous forme d'urticaire, d'éruption cutanée, de gonflements des yeux, de la bouche ou du visage, d'essoufflement ou de démangeaisons. Dans le cas de réactions allergiques sévères, des éruptions cutanées peuvent se présenter, lesquelles sont d'abord localisées, puis se développent sur tout le corps et sont liées à des desquamations généralisées (appelées syndrome de Stevens-Johnson et/ou nécrolyse épidermique toxique). Dans des cas très rares, outre les réactions cutanées, les réactions allergiques peuvent être accompagnées de fièvre, fatigue, gonflement des ganglions lymphatiques, d'une augmentation de certains globules blancs (éosinophilie) ainsi que d'effets sur le foie, les reins ou les poumons (appelées DRESS).
Imnovid ne doit pas être pris en même temps que des inhibiteurs puissants du CYP1A2 (p.ex. ciprofloxacine). Veuillez informer votre médecin si vous prenez des inhibiteurs puissants du CYP1A2 (p.ex. ciprofloxacine). Ces médicaments affectent le catabolisme d'Imnovid dans le corps. C'est pourquoi, si nécessaire, votre médecin ajustera votre dose d'Imnovid.
On a observé chez les patients traités par Imnovid une élévation des paramètres hépatiques dans le sang. Des cas d'inflammation du foie se sont également présentés, lesquels ont conduit à un arrêt du traitement. C'est pourquoi, votre médecin mesurera régulièrement votre fonction hépatique.
Compte tenu d'effets indésirables possibles comme fatigue, vertiges et confusion, vous devez être particulièrement prudent lorsque vous conduisez un véhicule ou utilisez des machines.
Il est important de noter que chez un petit nombre de patients atteints de myélome multiple, d'autres types de cancer peuvent se développer, et il est possible que ce risque augmente lors d'un traitement par Imnovid. C'est pourquoi, votre médecin évaluera soigneusement les bénéfices et risques avant de prescrire Imnovid.
De rares cas de réactivation de l'hépatite B ont été rapportés à la suite du traitement par Imnovid en association avec la dexaméthasone chez des patients présentant des antécédents d'infection par le virus de l'hépatite B. C'est pourquoi, votre médecin vous surveillera étroitement pendant le traitement par Imnovid afin de détecter les signes et symptômes d'une infection active par le virus de l'hépatite B. Veuillez informer votre médecin si vous avez des antécédents d'infection par le virus de l'hépatite B.
Ce médicament renferme moins de 1 mmol de sodium (23 mg) par gélule, ce qui signifie qu'il s'agit d'un médicament pratiquement «sans sodium».
Veuillez informer votre médecin ou votre pharmacien si vous souffrez d'une autre maladie, si vous êtes allergique ou si vous prenez d'autres médicaments (même en automédication!).
Imnovid peut-il être pris pendant la grossesse ou l’allaitement?
Imnovid ne doit pas être pris pendant une grossesse.
Les femmes en âge de procréer doivent absolument éviter de débuter une grossesse pendant le traitement par Imnovid. Pour exclure une grossesse, il convient d'effectuer des tests de grossesse avant, pendant et à la fin du traitement et d'utiliser des méthodes de contraception efficaces.
On ne sait pas si Imnovid passe dans le lait maternel. C'est pourquoi, Imnovid ne doit pas être administré pendant l'allaitement ou sinon, il convient d'arrêter l'allaitement.
Le pomalidomide passe dans le sperme humain. Les hommes dont la partenaire est en âge de procréer doivent utiliser un préservatif comme moyen de contraception pendant le traitement par Imnovid et pendant au moins 7 jours après l'arrêt du traitement.
Vous trouverez des informations détaillées concernant la contraception à la rubrique «Quelles sont les précautions à observer lors de la prise d'Imnovid?».
Comment utiliser Imnovid?
Prenez toujours Imnovid en suivant exactement les indications du médecin. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Prenez Imnovid à peu près à heure fixe chaque jour au cours ou en dehors d'un repas avec un peu d'eau. Les gélules ne doivent pas être cassées, ouvertes ou mâchées. Si la poudre d'une gélule ouverte de pomalidomide entre en contact avec la peau, lavez immédiatement et abondamment la peau au savon et à l'eau.
Pour sortir la gélule de la plaquette, appuyez seulement sur une extrémité de la gélule pour la pousser à travers la pellicule d'aluminium. N'exercez pas de pression sur le centre de la gélule car cela peut provoquer sa rupture.
Imnovid avec le bortézomib et la dexaméthasone
La dose recommandée d'Imnovid est de 4 mg une fois par jour. Prenez Imnovid pendant 14 jours consécutifs. La prise d'Imnovid est interrompue pendant les 7 jours suivants. Un cycle de traitement dure donc 21 jours. Répétez ensuite ce schéma d'utilisation jusqu'à ce que votre médecin vous demande d'arrêter la prise. Votre médecin déterminera la dose de bortézomib, l'un des médicaments qui vous sera administré en association avec Imnovid, en fonction de votre poids et de votre taille.
La dose de dexaméthasone, l'autre médicament que vous devez prendre en association avec Imnovid, est de 20 mg une fois par jour (ou de 10 mg une fois par jour si vous êtes âgé de plus de 75 ans), normalement conformément au schéma d'utilisation décrit dans le tableau suivant:
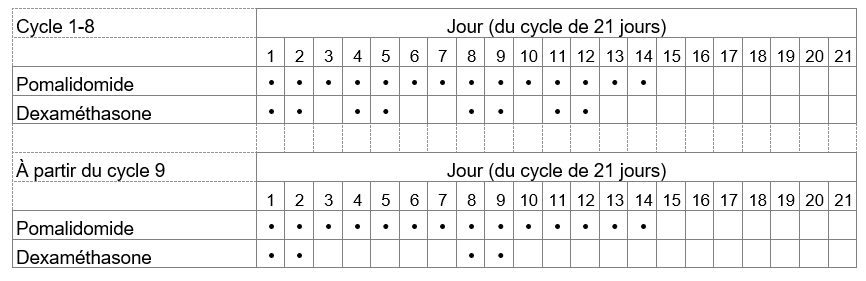
En fonction de l'appréciation de votre médecin, cette posologie pourra être ajustée au cas par cas.
Vous trouverez d'autres informations sur la dexaméthasone dans la notice d'emballage de la préparation utilisée.
Imnovid avec la dexaméthasone
La dose recommandée d'Imnovid est de 4 mg une fois par jour. Prenez Imnovid pendant 21 jours consécutifs. La prise d'Imnovid est interrompue pendant les 7 jours suivants. Un cycle de traitement dure donc 28 jours. Répétez ensuite ce schéma d'utilisation jusqu'à ce que votre médecin vous demande d'arrêter la prise.
La dose de dexaméthasone, le médicament que vous devez prendre en association avec Imnovid, est de 40 mg une fois par jour (ou de 20 mg une fois par jour si vous êtes âgé de plus de 75 ans). Le schéma d'utilisation habituel est le suivant: pendant chaque cycle de traitement de 28 jours, prenez la dexaméthasone aux jours 1, 8, 15 et 22 de chaque cycle. En fonction de l'appréciation de votre médecin, cette posologie pourra être ajustée au cas par cas.
Vous trouverez d'autres informations sur la dexaméthasone dans la notice d'emballage de la préparation utilisée.
Si vous subissez une dialyse en raison d'une insuffisance rénale, les jours de dialyse, vous devez prendre Imnovid après la dialyse.
Imnovid ne doit pas être utilisé chez les enfants et adolescents car il n'existe aucune étude sur cette tranche d'âge.
Informez immédiatement votre médecin si vous avez pris plus d'Imnovid que vous n'auriez dû.
Si vous avez oublié de prendre Imnovid un jour où vous auriez dû le prendre, prenez la prochaine gélule à l'heure habituelle le lendemain. Ne prenez pas de gélule supplémentaire pour compenser la dose d'Imnovid que vous avez oubliée de prendre la veille.
Ne changez pas de votre propre chef le dosage prescrit. Adressez-vous à votre médecin ou à votre pharmacien si vous estimez que l'efficacité du médicament est trop faible ou au contraire trop forte.
Quels effets secondaires Imnovid peut-il provoquer?
Très fréquents (concernent plus d'un utilisateur sur 10)
Infections, y compris infections des poumons, du nez, des sinus et de la gorge, diminution du nombre de globules blancs et rouges ainsi que des plaquettes (ce qui peut provoquer des infections, une fatigue et des saignements), taux faible de potassium dans le sang, taux élevé de sucre dans le sang, perte d'appétit, troubles du sommeil, neuropathies (p.ex. douleurs, brûlures, engourdissements ou sensation de faiblesse, fourmillements, perte du toucher, perte de la sensation de chaleur ou de douleur dans les membres ainsi que mouvements incontrôlés ou anormaux), vertiges, tremblements, essoufflement, toux, constipation, diarrhée, nausées, vomissements, crampes musculaires (spasmes ou douleurs), faiblesse musculaire, douleurs osseuses, douleurs dorsales, sensation de fatigue, fièvre, sensation de faiblesse, gonflement des membres (mollet, cheville) œdème généralisé comme p.ex. du visage.
Fréquents (concernent 1 à 10 utilisateurs sur 100)
Empoisonnement du sang, inflammation et gonflement des voies respiratoires dans les poumons, inflammation intestinale en association avec des douleurs abdominales et des diarrhées sanguinolentes, grippe, infections des voies urinaires, infections fongiques, infections fongiques dans la bouche, carcinome baso-cellulaire, sensation de soif, taux faible de sodium dans le sang, taux élevé de potassium dans le sang, taux élevé ou faible de calcium dans le sang, taux faible de phosphate dans le sang, taux faible d'albumine ou de magnésium dans le sang, augmentation du taux de l'ALT dans le sang (une valeur de la fonction hépatique), augmentation du taux de créatinine dans le sang (une valeur de la fonction rénale), confusion, anxiété, dépression, sautes d'humeur, perte de conscience courte et durable (syncope), somnolence, étourdissements, maux de tête, modification de la sensibilité gustative, vision trouble, opacification du cristallin de l'œil, modification de la fréquence ou du rythme cardiaque, accélération des battements du cœur, augmentation ou diminution de la pression artérielle, thromboses veineuses profondes (douleurs ou gonflements dans les jambes, en particulier dans le bas de la jambe ou le mollet), embolies pulmonaires (occlusion d'un vaisseau sanguin pulmonaire) et autres problèmes pulmonaires, nez bouché, mal de gorge, modification de la voix ou respiration sifflante, saignements y compris saignements de nez, dyspnée d'effort, maux de ventre, inflammation de la muqueuse buccale, lèvres sèches ou bouche sèche, ballonnement de l'estomac, éruptions cutanées, peau sèche, démangeaisons, sueurs excessives, douleurs dans les articulations ou les extrémités, douleurs dans le thorax, problèmes de mobilité, douleurs, insuffisance rénale, rétention urinaire, frissons, vertiges rotatoires, douleurs pelviennes, perte de poids, chute.
Occasionnels (concernent 1 à 10 utilisateurs sur 1000)
Coloration jaune de la peau et du blanc de l'œil (jaunisse).
Données de pharmacovigilance
Diminution du nombre de globules blancs et rouges et de plaquettes sanguines, syndrome de lyse tumorale et réactions allergiques graves (angiœdème, anaphylaxie, syndrome de Steven-Johnson, nécrolyse épidermique toxique, ou DRESS), élévation des paramètres hépatiques et inflammation du foie (p.ex. coloration foncée des urines, jaunisse), réactivation du virus de l'hépatite B, herpès zoster, hémorragies gastro-intestinales, carcinome épidermoïde de la peau, hypothyroïdie, maladie pulmonaire y compris inflammation des poumons.
Si vous remarquez des effets secondaires, veuillez en informer votre médecin ou votre pharmacien. Ceci vaut en particulier pour les effets secondaires non mentionnés dans cette notice d'emballage.
À quoi faut-il encore faire attention?
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques concernant le stockage
Conserver Imnovid dans l'emballage original, pas au-dessus de 25 °C et hors de la portée des enfants.
Remarques complémentaires
Rapportez toutes les gélules non utilisées ou endommagées à votre médecin ou à votre pharmacien pour qu'il en assure l'élimination correcte.
Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien, qui disposent d'une information détaillée destinée aux professionnels.
Que contient Imnovid?
Principes actifs
Pomalidomide: 1 mg, 2 mg, 3 mg ou 4 mg
Excipients
Mannitol, amidon prégélatinisé, fumarate de stéaryle sodique.
Enveloppe des gélules: gélatine, dioxyde de titane, oxyde de fer jaune (uniquement pour les gélules dures 1 mg, 2 mg et 3 mg), indigocarmine E132, érythrosine E127 (uniquement pour les gélules dures 2 mg), bleu brillant FCF E133 (uniquement pour les gélules dures 4 mg).
Encre d'impression: gomme laque, oxyde de fer noir, dioxyde de titane, Simeticon, propylène glycol, solution d'ammoniaque.
Numéro d’autorisation
61249 (Swissmedic).
Où obtenez-vous Imnovid? Quels sont les emballages à disposition sur le marché?
En pharmacie, seulement sur ordonnance médicale non renouvelable.
Imnovid 1 mg, 2 mg, 3 mg et 4 mg: chaque emballage contient 14 ou 21 gélules.
Titulaire de l’autorisation
Celgene GmbH, Zurich.
Cette notice d'emballage a été vérifiée pour la dernière fois en janvier 2020 par l'autorité de contrôle des médicaments (Swissmedic).
Che cos’è Imnovid e quando si usa?
Su prescrizione medica.
Imnovid contiene il principio attivo pomalidomide. Questo medicamento fa parte di un gruppo di medicamenti che agiscono sul funzionamento del sistema immunitario dell'organismo.
Imnovid viene usato nei pazienti affetti da mieloma multiplo (un tipo di tumore che provoca una forte proliferazione di alcune cellule nel midollo osseo) nel modo seguente:
- insieme al bortezomib (un cosiddetto inibitore del proteasoma) e al desametasone (un medicamento antinfiammatorio) nei pazienti che sono già stati sottoposti a una o più terapie,
- oppure solo insieme al desametasone, nei pazienti che sono già stati sottoposti a due o più terapie.
Quando non si può assumere Imnovid?
Se è in gravidanza o se pensa di esserlo, oppure se sta programmando una gravidanza.
Nelle donne potenzialmente fertili, a meno che non adottino rigorose misure contraccettive (vedere «Quando è richiesta prudenza nella somministrazione di Imnovid?»).
In caso di ipersensibilità al principio attivo pomalidomide, a una delle sostanze ausiliarie, alla thalidomide o alla lenalidomide.
Quando è richiesta prudenza nella somministrazione di Imnovid?
Per le donne che assumono Imnovid
Contraccezione
In casi particolari, il medico può prescrivere Imnovid a donne per cui esiste la possibilità di iniziare una gravidanza. Se questo è il suo caso, è assolutamente necessario seguire esattamente le istruzioni del medico. I punti seguenti sono molto importanti:
- Prima dell'inizio del trattamento, il suo medico verificherà che lei non sia in gravidanza e, inoltre, eseguirà un test di gravidanza ogni 4 settimane durante l'intero periodo del trattamento, incluse le sospensioni temporanee, e 4 settimane dopo la fine del trattamento.
- Il suo medico o un altro medico le fornirà consulenza in merito ai metodi contraccettivi adatti. Dovrà iniziare tali misure contraccettive in genere 4 settimane prima di iniziare il trattamento e attuarle rigorosamente non solo per tutta la durata del trattamento, incluse le sospensioni temporanee, ma anche per 4 settimane dopo la fine del trattamento.
- Nel caso in cui, nonostante il rigoroso rispetto delle misure contraccettive, lei inizi una gravidanza o presuma di poter essere in gravidanza durante il trattamento con Imnovid o nel mese successivo alla fine del trattamento deve informarne immediatamente il suo medico. In tal caso il suo medico adotterà le misure necessarie.
Per gli uomini che assumono Imnovid
I pazienti di sesso maschile che hanno rapporti sessuali con una donna in età fertile devono usare preservativi per tutta la durata del trattamento, incluse le sospensioni temporanee, e per 7 giorni dopo la fine del trattamento. Inoltre, durante il trattamento con Imnovid e per i 7 giorni successivi non devono donare lo sperma.
A causa del rischio per il nascituro, non deve mai dare Imnovid ad altre persone.
Il titolare dell'omologazione di Imnovid metterà a disposizione il materiale seguente:
- informazioni relative ai problemi per la gravidanza;
- opuscolo per i pazienti con un questionario, che dovrà firmare, per confermare di avere compreso la necessità di evitare una gravidanza durante la terapia con Imnovid.
Altre precauzioni
Prima e durante il trattamento con Imnovid, il suo medico la sottoporrà a regolari analisi del sangue, poiché questo medicamento può causare una riduzione del numero delle cellule del sangue che contrastano le infezioni e di quelle che sono responsabili della coagulazione.
Sulla base dei risultati delle analisi del sangue e del suo stato di salute generale, il medico potrà eventualmente modificare la dose di Imnovid o interrompere il trattamento.
Durante il trattamento con Imnovid aumenta il rischio di formazione di coaguli di sangue nelle vene (tromboembolia venosa). Si rivolga immediatamente al suo medico se dovessero comparire i seguenti sintomi: febbre, brividi, mal di gola, tosse, ulcere della bocca o altri sintomi di infezione, sanguinamento o lividi senza una causa riconoscibile, dolore alle gambe o al petto, fiato corto.
Nei pazienti con malattia cardiaca preesistente o fattori di rischio cardiaco può manifestarsi insufficienza cardiaca. Si rivolga al suo medico se ha avuto un attacco di cuore, ha insufficienza cardiaca, ha difficoltà di respirazione, o se fuma, ha la pressione sanguigna alta o alti livelli di colesterolo.
Soprattutto nei pazienti che prima del trattamento con Imnovid presentano un imponente carico tumorale, la degradazione rapida delle cellule cancerose può causare una cosiddetta sindrome da lisi tumorale. Se osserva sintomi quali nausea, affanno respiratorio, battito cardiaco irregolare, urina torbida, stanchezza e/o disturbi articolari, si rivolga al suo medico.
Eventuali reazioni allergiche gravi (angioedema e anafilassi) possono manifestarsi sotto forma di orticaria, eruzioni cutanee, gonfiore a carico di occhi, bocca o viso, affanno respiratorio o prurito. In caso di reazioni allergiche gravi possono manifestarsi eruzioni cutanee che in principio sono localmente circoscritte e, successivamente, possono estendersi all'intero corpo ed essere associate a desquamazione di ampie aree cutanee (sindrome di Stevens-Johnson e/o necrolisi epidermica tossica). In casi molto rari, le reazioni allergiche possono essere accompagnate, oltre che da reazioni cutanee, da febbre, stanchezza, ingrossamento dei linfonodi, aumento di determinati globuli bianchi (eosinofilia), nonché da effetti su fegato, reni o polmoni (una reazione denominata DRESS).
Imnovid non deve essere assunto in concomitanza con potenti inibitori del CYP1A2 (per es. ciprofloxacina). Informi il suo medico, se assume potenti inibitori del CYP1A2 (per es. ciprofloxacina). Questi medicamenti influenzano la degradazione di Imnovid nell'organismo, per cui il suo medico adeguerà la sua dose di Imnovid a seconda del bisogno.
Nei pazienti trattati con Imnovid è stato osservato un aumento dei valori di funzionalità epatica nel sangue. Vi sono stati anche casi di epatite, che hanno portato a un'interruzione del trattamento. Per questo motivo, il suo medico eseguirà valutazioni periodiche della funzionalità epatica.
A causa di possibili effetti collaterali, quali stanchezza, capogiro e confusione mentale, deve essere particolarmente prudente nella guida di veicoli o nell'uso di macchinari.
È importante tenere presente che in un piccolo numero di pazienti affetti da mieloma multiplo possono svilupparsi ulteriori forme tumorali ed è possibile che questo rischio aumenti in caso di trattamento con Imnovid. Per questo motivo, prima di prescrivere Imnovid il suo medico valuterà in maniera approfondita il rapporto rischi-benefici.
In un piccolo numero di pazienti che erano stati precedentemente infettati con il virus dell'epatite B è stata osservata una riattivazione dell'epatite B nel corso di un trattamento con Imnovid in combinazione con desametasone. Per questo motivo, durante la terapia con Imnovid il suo medico la esaminerà attentamente per escludere eventuali segni e sintomi di un'infezione attiva da virus dell'epatite B. Informi il suo medico, se in passato ha contratto un'infezione da virus dell'epatite B.
Questo medicamento contiene meno di 1 mmol di sodio (23 mg) per capsula, ovvero è quasi «privo di sodio».
Informi il suo medico o il suo farmacista nel caso in cui soffra di altre malattie, soffra di allergie o assuma o applichi esternamente altri medicamenti (anche se acquistati di sua iniziativa!).
Si può assumere Imnovid durante la gravidanza o l’allattamento?
Imnovid non deve essere usato durante la gravidanza.
Le donne in età fertile non devono iniziare una gravidanza durante il trattamento con Imnovid. Al fine di escludere una gravidanza, prima, durante e alla fine del trattamento devono essere eseguiti test di gravidanza e devono essere adottati metodi contraccettivi efficaci.
Non è noto se Imnovid passi nel latte materno. Pertanto, Imnovid non deve essere usato durante l'allattamento, oppure l'allattamento deve essere interrotto.
La pomalidomide passa nel liquido seminale umano. I pazienti di sesso maschile con una partner in età fertile devono usare preservativi a scopo contraccettivo durante il trattamento con Imnovid e per almeno 7 giorni dopo la fine del trattamento.
Indicazioni dettagliate riguardo alla contraccezione sono riportate nel paragrafo «Quando è richiesta prudenza nella somministrazione di Imnovid?».
Come usare Imnovid?
Prenda Imnovid seguendo sempre esattamente le istruzioni del suo medico. Se ha dubbi, consulti il suo medico o il suo farmacista.
Prenda Imnovid ogni volta circa alla stessa ora, indipendentemente dai pasti, con un po' d'acqua. Le capsule non devono essere spezzate, aperte o masticate. Se la polvere di una capsula spezzata di pomalidomide viene a contatto con la cute, lavi subito accuratamente la zona interessata con acqua e sapone.
Per estrarre la capsula dal blister, fare pressione su un solo lato della capsula, spingendola attraverso il foglio d'alluminio. Non premere sul centro della capsula, altrimenti si rischia di romperla.
Imnovid con bortezomib e desametasone
La dose raccomandata di Imnovid è di 4 mg una volta al giorno. Prenda Imnovid per 14 giorni consecutivi. Per i 7 giorni successivi, l'assunzione di Imnovid viene sospesa. Un ciclo di trattamento dura perciò 21 giorni. Ripeta questo schema di assunzione fino a quando il suo medico non le chiede di interrompere il trattamento.
Il suo medico stabilirà la dose del bortezomib, il medicamento da assumere in associazione a Imnovid, sulla base del suo peso e della sua altezza.
La dose del desametasone, l'altro medicamento da assumere in associazione a Imnovid, è di 20 mg una volta al giorno (o 10 mg una volta al giorno, se ha più di 75 anni di età), di solito secondo lo schema di assunzione indicato nella tabella seguente:
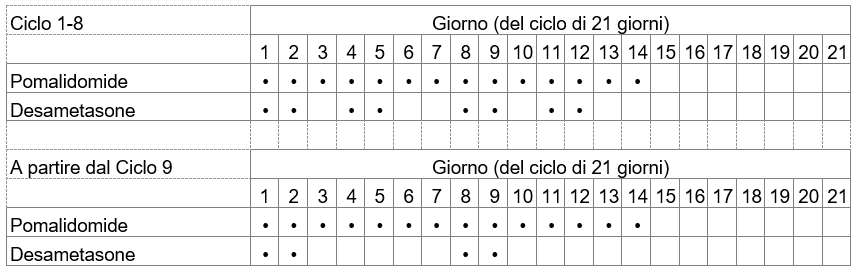
Questa posologia può essere modificata individualmente a giudizio del suo medico.
Altre indicazioni sul desametasone sono riportate nel foglietto illustrativo del relativo medicamento.
Imnovid insieme al desametasone
La dose raccomandata di Imnovid è di 4 mg una volta al giorno. Prenda Imnovid per 21 giorni consecutivi. Per i 7 giorni successivi, l'assunzione di Imnovid viene sospesa. Un ciclo di trattamento dura perciò 28 giorni. Ripeta questo schema di assunzione fino a quando il suo medico non le chiede di interrompere il trattamento.
La dose di desametasone, il medicamento da assumere in associazione a Imnovid, è di 40 mg una volta al giorno (o 20 mg una volta al giorno, se ha più di 75 anni di età). Lo schema di assunzione è di solito il seguente: durante il ciclo di trattamento di 28 giorni prenda il desametasone i giorni 1, 8, 15 e 22 di ogni ciclo. A giudizio del suo medico, questa posologia può essere modificata individualmente.
Altre indicazioni sul desametasone sono riportate nel foglietto illustrativo del relativo medicamento.
Se viene sottoposto a dialisi a causa di insufficienza renale, nei giorni di dialisi deve assumere Imnovid solo dopo la procedura dialitica.
Imnovid non viene usato nei bambini e negli adolescenti, perché non esistono indagini scientifiche in queste fasce di età.
Informi immediatamente il suo medico se ha preso una quantità di Imnovid superiore a quella prescritta.
Se dimentica di prendere Imnovid nel giorno prescritto, prenda la capsula successiva alla solita ora il giorno successivo. Non aumenti il numero di capsule da assumere per compensare la dimenticanza della dose di Imnovid il giorno precedente.
Non modifichi di propria iniziativa la posologia prescritta. Se ritiene che l'azione del medicamento sia troppo debole o troppo forte ne parli al suo medico o al suo farmacista.
Quali effetti collaterali può avere Imnovid?
Molto comune (riguarda più di 1 utilizzatore su 10)
Infezioni, incluse infezioni dei polmoni, del naso, dei seni paranasali e della gola, riduzione del numero di globuli bianchi e rossi nonché di piastrine (che può causare infezioni, stanchezza ed emorragie), bassi livelli di potassio nel sangue, alti livelli di zucchero nel sangue, mancanza di appetito, disturbi del sonno, neuropatie (ad es. dolore, bruciore, sensazione di intorpidimento o di debolezza, formicolio, perdita della sensibilità al tatto, al calore o al dolore negli arti e movimenti incontrollati o anomali), capogiri, tremore, respiro corto, tosse, stitichezza, diarrea, nausea, vomito, crampi muscolari (spasmo o dolore), debolezza muscolare, dolore alle ossa, mal di schiena, sensazione di stanchezza, febbre, senso di debolezza, gonfiore agli arti (polpacci, caviglie), gonfiore generale, ad es. del viso.
Comune (riguarda da 1 a 10 utilizzatori su 100)
Intossicazione del sangue, infiammazione e gonfiore delle vie respiratorie nei polmoni, infiammazione intestinale associata a dolore al basso ventre e diarrea sanguinolenta, influenza, infezioni delle vie urinarie, infezioni da funghi, infezioni da funghi alla bocca, carcinoma basocellulare, sensazione di sete, bassi livelli di sodio nel sangue, alti livelli di potassio nel sangue, alti o bassi livelli di calcio nel sangue, bassi livelli di fosfato nel sangue, bassi livelli di albumina o magnesio nel sangue, aumento dei livelli di ALT nel sangue (un valore della funzionalità del fegato), aumento dei livelli di creatinina nel sangue (un valore della funzionalità del rene), confusione, stati d'ansia, depressione, sbalzi d'umore, perdita di coscienza di breve durata (sincope), sonnolenza, stordimento, mal di testa, alterazione del gusto, vista offuscata, opacità del cristallino, alterazioni della frequenza o del ritmo cardiaco, battito cardiaco accelerato, pressione sanguigna alta o bassa, trombosi venosa profonda (dolore o gonfiore agli arti inferiori, in particolare alla gamba o al polpaccio), embolia polmonare (ostruzione di un vaso sanguigno nei polmoni) e altri problemi polmonari, naso chiuso, mal di gola, alterazioni della voce o ronchi, sanguinamenti (incluso sanguinamento nasale), dispnea da sforzo, dolore addominale, infiammazione della mucosa orale, secchezza delle labbra o della bocca, distensione dell'addome, eruzioni cutanee, secchezza della cute, prurito, aumento della sudorazione, dolore alle articolazioni o alle estremità, dolore al torace, difficoltà di movimento, dolori, insufficienza renale, ritenzione urinaria, brividi, vertigini rotatorie, dolore al bacino, perdita di peso, caduta.
Non comune (riguarda da 1 a 10 utilizzatori su 1000)
Ingiallimento della pelle e del bianco degli occhi (ittero).
Esperienze successive all'omologazione
Riduzione del numero di globuli bianchi e rossi e di piastrine, sindrome da lisi tumorale, reazioni allergiche gravi (angioedema, anafilassi, sindrome di Steven-Johnson, necrolisi epidermica tossica o DRESS), aumento dei valori di funzionalità epatica ed epatite (ad es. urine scure, ittero), riattivazione del virus dell'epatite B, herpes zoster, emorragie gastrointestinali, carcinoma spinocellulare della cute, ipotiroidismo, malattie polmonari, inclusa polmonite.
Se osserva effetti collaterali, si rivolga al suo medico o farmacista, soprattutto se si tratta di effetti collaterali non descritti in questo foglietto illustrativo.
Di che altro occorre tener conto?
Il medicamento non deve essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Indicazioni per la conservazione
Conservi Imnovid nella confezione originale, a temperatura non superiore ai 25 °C e fuori dalla portata dei bambini.
Altre indicazioni
Porti le capsule non utilizzate o danneggiate al suo medico o al suo farmacista per uno smaltimento corretto.
Il medico o il farmacista, che sono in possesso di un'informazione professionale dettagliata, possono darle ulteriori informazioni.
Cosa contiene Imnovid?
Principi attivi
Pomalidomide: 1 mg, 2 mg, 3 mg o 4 mg
Sostanze ausiliarie
Mannitolo, amido pregelatinizzato, stearil fumarato di sodio.
Rivestimento della capsula: gelatina, biossido di titanio, ossido di ferro giallo (solo per capsule rigide 1 mg, 2 mg e 3 mg), indigocarmina E132, eritrosina E127 (solo per capsule rigide 2 mg), blu brillante FCF E133 (solo per capsule rigide 4 mg).
Inchiostro della dicitura: gommalacca, ossido di ferro nero, biossido di titanio, simeticon, glicole propilenico, soluzione di ammoniaca.
Numero dell’omologazione
61249 (Swissmedic).
Dove è ottenibile Imnovid? Quali confezioni sono disponibili?
In farmacia dietro presentazione della ricetta medica non rinnovabile.
Imnovid 1 mg, 2 mg, 3 mg e 4 mg: confezioni da 14 o 21 capsule rigide
Titolare dell’omologazione
Celgene GmbH, Zurigo.
Questo foglietto illustrativo è stato controllato l'ultima volta nel gennaio 2020 dall'autorità competente in materia di medicamenti (Swissmedic).
Zusammensetzung
Wirkstoffe
Pomalidomidum.
Hilfsstoffe
Mannitol, vorverkleisterte Stärke, Natriumstearylfumarat.
Kapselhülle: Gelatine, Titandioxid, gelbes Eisenoxid (nur bei Hartkapseln 1 mg, 2 mg und 3 mg), Indigocarmin E132, Erythrosin E127 (nur bei Hartkapseln 2 mg), Brillantblau FCF E133 (nur bei Hartkapseln 4 mg).
Drucktinte: Shellack, schwarzes Eisenoxid, Titandioxid, Simeticon, Propylenglycol, Ammoniak-Lösung.
Eine Hartkapsel enthält max. 0,018 mg (Hartkapseln 1 mg) bzw. max. 0,036 mg (Hartkapseln 2 mg) bzw. max. 0,025 mg (Hartkapseln 3 mg) bzw. max. 0,033 (Hartkapseln 4 mg) Natrium.
Darreichungsform und Wirkstoffmenge pro Einheit
Hartkapseln zu 1 mg, 2 mg, 3 mg und 4 mg.
Indikationen/Anwendungsmöglichkeiten
Imnovid in Kombination mit Bortezomib und Dexamethason ist indiziert zur Behandlung von erwachsenen Patienten mit multiplem Myelom (MM), welche mindestens eine vorgängige Therapie, inklusive Lenalidomid, erhielten.
Imnovid in Kombination mit Dexamethason ist indiziert zur Behandlung von rezidiviertem und refraktärem multiplem Myelom bei Patienten, welche mindestens zwei vorgängige Therapien erhielten (inklusive Lenalidomid und Bortezomib) und welche eine Progredienz zur letzten Therapie gezeigt haben.
Dosierung/Anwendung
Die Behandlung muss von einem erfahrenen Hämatologen oder Onkologen begonnen und überwacht werden.
Imnovid in Kombination mit Bortezomib und Dexamethason (PVd) bei Patienten mit multiplem Myelom, welche mindestens eine vorgängige Therapie erhalten haben
Die empfohlene Anfangsdosis von Imnovid beträgt 4 mg einmal täglich oral an den Tagen 1-14 der sich wiederholenden 21-tägigen Behandlungszyklen.
Die empfohlene Dosis von Bortezomib beträgt 1,3 mg/m2 und die empfohlene Dosis von Dexamethason beträgt 20 mg/Tag oral einmal täglich, wobei die Anwendung entsprechend dem in Tabelle 1 gezeigten Dosierungsschema erfolgt.
Die Dosierung wird auf der Basis von klinischen Befunden und Laborbefunden fortgesetzt oder modifiziert. Die Behandlung soll bei Fortschreiten der Erkrankung abgebrochen werden.
Tabelle 1: Empfohlenes Dosierungsschema für Imnovid in Kombination mit Bortezomib und Dexamethason
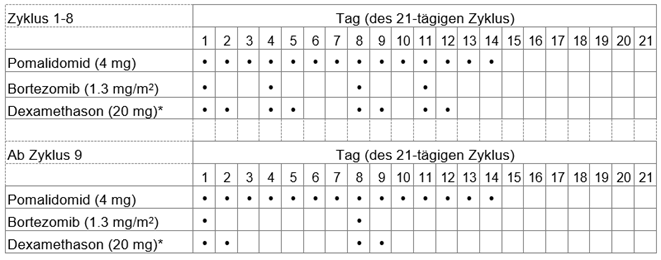
* Für Patienten >75 Jahre siehe Abschnitt «Spezielle Dosierungsanweisungen».
Imnovid in Kombination mit Dexamethason (Pd) bei Patienten mit rezidiviertem und refraktärem multiplem Myelom, welche mindestens zwei vorgängige Therapien erhalten haben
Die empfohlene Anfangsdosis von Imnovid beträgt 4 mg einmal täglich oral an den Tagen 1-21 der sich wiederholenden 28-tägigen Behandlungszyklen bis zur Progredienz. Die empfohlene Dosis Dexamethason beträgt 40 mg einmal täglich oral an den Tagen 1, 8, 15 und 22 eines jeden 28‑Tage-Behandlungszyklus.
Die Dosierung wird auf der Basis von klinischen Befunden und Laborbefunden fortgesetzt oder modifiziert.
Das Dosierungsschema ist in Tabelle 2 gezeigt.
Tabelle 2: Empfohlenes Dosierungsschema für Imnovid in Kombination mit Dexamethason
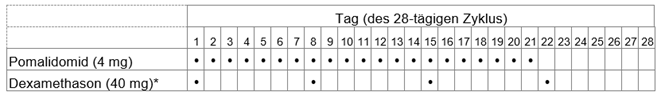
* Für Patienten >75 Jahre siehe Abschnitt «Spezielle Dosierungsanweisungen».
Dosisanpassung oder -unterbrechung
Hämatotoxizität
Bei einer Thrombozytopenie mit Abfall der Werte auf <25 x 109/l oder bei einer Neutropenie mit Abfall der Werte auf <0,5 x 109/l oder bei einer febrilen Neutropenie (Fieber ≥38,5 °C und ANZ <1,0 x 109/l) sollte die Behandlung mit Pomalidomid unterbrochen werden, gefolgt von wöchentlichen Kontrollen des vollständigen Blutbildes (und im Fall des Absinkens der Neutrophilenzahl zusätzlich G-CSF im Ermessen des behandelnden Arztes). Nach Normalisierung der Thrombozyten/Neutrophilenzahlen sollte die Behandlung mit Pomalidomid in einer Dosierung von 3 mg täglich fortgeführt werden. Bei jedem nachfolgenden Abfall (<25 x 109/l respektive <0,5 × 109/l) sollte die Behandlung mit Pomalidomid unterbrochen werden. Nach Normalisierung der Thrombozyten/Neutrophilenzahlen sollte die Behandlung mit Pomalidomid in einer gegenüber der letzten Dosis um 1 mg reduzierten Dosierung fortgeführt werden.
Imnovid in Kombination mit Bortezomib und Dexamethason (PVd): Um einen neuen Pomalidomid-Zyklus beginnen zu können, muss die Neutrophilenzahl ≥1 × 109/l und die Thrombozytenzahl ≥50 × 109/l sein.
Imnovid in Kombination mit Dexamethason (Pd): Um einen neuen Pomalidomid-Zyklus beginnen zu können, muss die Neutrophilenzahl ≥0,5 × 109/l und die Thrombozytenzahl ≥50 × 109/l sein.
Weitere Toxizitäten 3./4. Grades
Bei anderen Grad 3/4 Toxizitäten, bei denen ein Zusammenhang mit Pomalidomid angenommen wird, ist die Behandlung abzusetzen und nach ärztlichem Ermessen in einer gegenüber der letzten Dosis um 1 mg reduzierten Dosierung wiederaufzunehmen, wenn sich die Toxizität auf ≤ Grad 2 zurückgebildet hat. Wenn die jeweiligen Toxizitäten auch nach Dosisreduktion auf 1 mg auftreten, muss das Arzneimittel abgesetzt werden.
Für Dosisanpassungen aufgrund der Toxizität von Bortezomib wird auf die entsprechende Arzneimittel-Fachinformation verwiesen.
Dosisanpassung bei gleichzeitiger Gabe von CYP1A2-Hemmern
Vermeiden Sie die gleichzeitige Anwendung von Pomalidomid mit starken CYP1A2-Hemmern. Ziehen Sie alternative Behandlungsmethoden in Betracht. Wenn starke CYP1A2-Hemmer (z.B. Ciprofloxacin und Fluvoxamin) gleichzeitig mit Pomalidomid verabreicht werden, muss die Pomalidomid-Dosis um 50% reduziert werden.
Absetzen von Pomalidomid
Bei einem Grad-2- oder Grad-3-Hautausschlag sollte eine Unterbrechung oder das Absetzen der Behandlung mit Pomalidomid erwogen werden.
Bei Angioödem, Anaphylaxie, Grad-4-Hautausschlag, exfoliativem oder bullösem Hautausschlag oder bei Verdacht auf Stevens-Johnson-Syndrom (SJS), toxischer epidermaler Nekrolyse (TEN) oder Arzneimittelexanthem mit Eosinophilie und systemischen Symptomen (DRESS) muss Pomalidomid abgesetzt werden. Wurde die Behandlung wegen derartiger Reaktionen beendet, sollte sie nicht mehr aufgenommen werden.
Spezielle Dosierungsanweisungen
Patienten mit Leberfunktionsstörungen
Für Patienten mit leicht, bis mässig eingeschränkter Leberfunktion (Child-Pugh-Klassen A oder B) beträgt die empfohlene Anfangsdosis 3 mg täglich (Dosisreduktion um 25%). Für Patienten mit stark eingeschränkter Leberfunktion (Child-Pugh Klasse C) beträgt die empfohlene Dosis 2 mg (Dosisreduktion um 50%).
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit mässiger Niereninsuffizienz und Patienten mit schwerer Niereninsuffizienz, die nicht dialysepflichtig sind, ist keine Dosisanpassung von Pomalidomid erforderlich. Bei dialysepflichtigen Patienten mit schwerer Niereninsuffizienz beträgt die empfohlene Anfangsdosis 3 mg täglich (Dosisreduktion um 25%). An Hämodialysetagen sollte Pomalidomid nach der Hämodialyse eingenommen werden.
Ältere Patienten
Für Pomalidomid ist keine Dosierungsanpassung erforderlich.
Imnovid in Kombination mit Bortezomib und Dexamethason (PVd) nach mindestens einer vorgängigen Therapie:
Bei Patienten >75 Jahre beträgt die Dexamethason-Dosis 10 mg einmal täglich an den Tagen 1, 2, 4, 5, 8, 9, 11 und 12 eines 21-tägigen Zyklus für die Zyklen 1-8, und von Zyklus 9 an 10 mg einmal täglich an den Tagen 1, 2, 8 und 9 eines 21-tägigen Zyklus.
Imnovid in Kombination mit Dexamethason (Pd) nach mindestens 2 vorgängigen Therapien:
Bei Patienten >75 Jahre beträgt die Anfangsdosis von Dexamethason 20 mg einmal täglich an den Tagen 1, 8, 15 und 22 eines jeden 28‑Tage-Zyklus.
Kinder und Jugendliche
Pomalidomid wurde bei pädiatrischen Patienten nicht untersucht.
Art der Anwendung
Imnovid sollte jeden Tag etwa zur gleichen Tageszeit oral eingenommen werden. Die Kapseln sollen nicht geöffnet, zerbrochen oder zerkaut werden. Die Imnovid-Kapseln sollen unabhängig von einer Mahlzeit vorzugsweise mit etwas Wasser unzerkaut eingenommen werden. Wenn der Patient die Einnahme von Imnovid an einem Tag vergessen hat, sollte er die nächste Kapsel zur gewohnten Zeit am nächsten Tag einnehmen. Patienten sollen die Zahl der eingenommenen Kapseln nicht erhöhen, um die vergessene Dosis Imnovid vom Vortag nachzuholen.
Zur Anwendung anderer Arzneimittel in Kombination mit Pomalidomid wird auf die entsprechende Fachinformation für das jeweilige Arzneimittel verwiesen.
Kontraindikationen
Überempfindlichkeit gegenüber Pomalidomid oder einem der Hilfsstoffe oder Thalidomid und Lenalidomid.
Schwangerschaft.
Gebärfähige Frauen, ausser wenn alle Bedingungen des Schwangerschaftsverhütungsprogramms erfüllt sind.
Zur Anwendung anderer Arzneimittel in Kombination mit Pomalidomid wird auf die entsprechende Fachinformation für das jeweilige Arzneimittel verwiesen.
Warnhinweise und Vorsichtsmassnahmen
Pomalidomid ist ein Thalidomidanalog. Thalidomid hat bekanntlich reproduktionstoxische Wirkungen beim Menschen, die zu schweren lebensbedrohlichen Geburtsfehlern führen. Pomalidomid hat sich sowohl bei Ratten als auch bei Kaninchen als reproduktionstoxisch erwiesen, wenn es in der Phase gegeben wird, in der die wichtigen Organe angelegt werden (siehe «Präklinische Daten»). Wird Pomalidomid während der Schwangerschaft eingenommen, ist auch beim Menschen mit einer reproduktionstoxischen Wirkung zu rechnen.
Schwangerschaftsverhütungsprogramm
Programm bei Patientinnen
Die Bedingungen des Schwangerschaftsverhütungsprogramms müssen bei allen Patientinnen erfüllt sein, ausser wenn die Patientin erwiesenermassen nicht schwanger werden kann.
Kriterien zur Abklärung des Schwangerschaftspotentials
Eine Patientin oder Partnerin eines männlichen Patienten wird als gebärfähig klassifiziert, ausser sie erfüllt mindestens eine der folgenden Bedingungen:
- Alter ≥50 Jahre und spontan amenorrhoisch während ≥1 Jahr*
- Bestätigtes vorzeitiges Ovarialversagen
- Vorhergehende beidseitige Salpingo-Oophorektomie, Tubensterilisation oder Hysterektomie
- XY-Genotyp, Turner Syndrom, Uterus-Aplasie
* Eine Amenorrhoe nach Krebstherapie schliesst Gebärfähigkeit nicht aus.
Beratung
Bei gebärfähigen Frauen ist Pomalidomid kontraindiziert, wenn nicht alle der folgenden Bedingungen erfüllt sind:
- Die Patientin versteht das zu erwartende teratogene Risiko für das ungeborene Kind.
- Sie versteht die Notwendigkeit einer wirksamen Schwangerschaftsverhütung ohne Unterbrechung 4 Wochen vor Behandlungsbeginn, während der ganzen Behandlungsdauer inklusive Behandlungsunterbrüchen und 4 Wochen nach Beendigung der Behandlung.
- Sogar wenn eine gebärfähige Patientin amenorrhoisch ist, muss sie alle Empfehlungen zu einer wirksamen Kontrazeption befolgen.
- Sie soll fähig sein, sich an wirksame kontrazeptive Massnahmen zu halten.
- Sie ist informiert und versteht die Konsequenzen einer Schwangerschaft und die Notwendigkeit, rasch medizinischen Rat zu suchen, falls eine Schwangerschaft vermutet wird.
- Sie versteht die Notwendigkeit und ist bereit, Schwangerschaftstests alle 4 Wochen durchführen zu lassen.
- Sie hat bestätigt, dass sie die Gefahren und notwendigen Vorsichtsmassnahmen im Zusammenhang mit der Einnahme von Pomalidomid verstanden hat.
Der verschreibende Arzt muss bei gebärfähigen Frauen sicherstellen, dass
- die Patientin die obenstehenden Bedingungen erfüllt.
- die Patientin die Bedingungen zur Schwangerschaftsverhütung einhält, einschliesslich der Bestätigung eines genügenden Verständnisses.
- die Patientin ausreichende kontrazeptive Massnahmen während mindestens 4 Wochen vor Beginn der Behandlung angewendet hat und wirksame kontrazeptive Massnahmen während der ganzen Behandlungszeit inklusive Behandlungsunterbrüchen und während mindestens 4 Wochen nach Ende der Behandlung weiterführen wird. Bei Patientinnen, bei welchen eine sofortige Behandlung mit Pomalidomid notwendig ist, muss eine adäquate Kontrazeption inkl. Verwendung von Kondomen während 7 Tagen vor Beginn der Behandlung durchgeführt werden.
- ein negatives Resultat eines Schwangerschaftstests vor Beginn der Behandlung vorliegt.
Kontrazeption
Gebärfähige Frauen müssen während 4 Wochen vor Beginn der Behandlung, während der ganzen Behandlungszeit inkl. Behandlungsunterbrüchen und während 4 Wochen nach Abschluss der Behandlung wirksame kontrazeptive Methoden anwenden. Bei Patientinnen, bei denen eine sofortige Behandlung mit Pomalidomid notwendig ist, muss während 7 Tagen vor Beginn der Behandlung eine wirksame Kontrazeption inkl. Verwendung von Kondomen durchgeführt werden. Falls nicht schon vorher wirksame kontrazeptive Methoden angewendet wurden, muss die Patientin an eine medizinische Beratungsstelle überwiesen werden, wo sie eine umfassende Beratung betreffend wirksamer kontrazeptiver Methoden erhält.
Die folgenden Verfahren können als wirksame kontrazeptive Methoden angesehen werden:
- von der Patientin unabhängige Methoden:
- Implantat
- Medroxyprogesteron-Acetat-Depot
- Sterilisation
- von der Patientin abhängige Methoden:
- Abstinenz von heterosexuellem Geschlechtsverkehr
- Heterosexueller Geschlechtsverkehr nur mit einem vasektomierten männlichen Partner; die Vasektomie muss durch zweimalige negative Spermauntersuchung bestätigt werden
- Orale Kontrazeptiva nur Progesteron enthaltend.
Wegen des erhöhten Risikos venöser Thromboembolien unter Pomalidomid werden kombinierte orale Kontrazeptiva nicht empfohlen. Falls eine Patientin bereits kombinierte orale Kontrazeptiva verwendet, sollte ein Wechsel zu einer anderen kontrazeptiven Methode in Betracht gezogen werden. Das Risiko venöser Thromboembolien bleibt während 4-6 Wochen nach Abschluss der Behandlung mit kombinierten oralen Kontrazeptiva bestehen. Falls andere Methoden nicht angewendet werden können, sollte eine Thromboseprophylaxe während der weiteren Verwendung der kombinierten oralen Kontrazeptiva in Betracht gezogen werden. Die Patientin sollte angemessen über das Risiko einer venösen Thromboembolie informiert werden.
Intrauterine Systeme haben ein erhöhtes Risiko von Infektionen beim Einsetzen und können zu unregelmässigen vaginalen Blutungen führen. Diese Methoden werden daher nicht empfohlen.
Schwangerschaftstests
Es müssen Schwangerschaftstests mit einer Empfindlichkeit von mindestens 25 IU/mL hCG bei gebärfähigen Frauen durchgeführt werden.
Jeder Fall einer Patientin mit einem positiven Schwangerschaftstest muss unverzüglich dem Swiss Teratogen Information Service (STIS), Lausanne mit dem Swissmedic-Formular «Meldung einer vermuteten unerwünschten Arzneimittelwirkung (UAW)» gemeldet werden.
- Vor Beginn einer Behandlung
Ein Schwangerschaftstest muss während der Konsultation, bei welcher Pomalidomid verschrieben wird, oder innerhalb von drei Tagen vor dem Besuch des verschreibenden Arztes durchgeführt werden, nachdem die Patientin während mindestens 4 Wochen eine wirksame Kontrazeption durchgeführt hat. Der Test soll sicherstellen, dass die Patientin bei Beginn der Behandlung mit Pomalidomid nicht schwanger ist.
- Vor Beginn der Behandlung, wenn sofortige Behandlung notwendig ist
Ein quantitativer hCG-Test im Serum sollte sofort durchgeführt werden. Nach wirksamer Kontrazeption inkl. Verwendung eines Kondoms während 7 Tagen muss dieser Test wiederholt werden. Falls beide Tests bestätigen, dass die Patientin nicht schwanger ist, kann mit der Behandlung begonnen werden.
- Während und bei Abschluss der Behandlung
Ein Schwangerschaftstest muss alle 4 Wochen, einschliesslich 4 Wochen nach Abschluss der Behandlung, wiederholt werden. Diese Schwangerschaftstests sollten während der Arztbesuche zur Verschreibung von Pomalidomid oder in den drei Tagen vor dem Arztbesuch durchgeführt werden.
Am besten sollten Schwangerschaftstests, Verschreibung und Abgabe von Pomalidomid am gleichen Tag erfolgen. Die Abgabe von Pomalidomid muss innerhalb von maximal 7 Tagen nach der Verschreibung erfolgen.
Programm bei Patienten
Klinische Daten belegen, dass es bei männlichen Patienten während der Einnahme von Imnovid zum Übertritt dieses Wirkstoffs in das Sperma kommt. Patienten mit gebärfähigen Partnerinnen sollten deshalb während der Behandlung mit Imnovid und mindestens für 7 Tage nach Beendigung der Behandlung beim Geschlechtsverkehr Kondome benutzen. Männer, welche Imnovid einnehmen, müssen folgende Bedingungen erfüllen:
- Sie müssen das zu erwartende teratogene Risiko verstehen, falls sie mit einer gebärfähigen Frau Geschlechtsverkehr haben.
- Sie müssen verstehen und damit einverstanden sein, während der ganzen Behandlungsdauer, inklusive Behandlungsunterbrüchen und während 7 Tagen nach Abschluss der Behandlung ein Kondom zu benützen, wenn sie mit einer schwangeren oder gebärfähigen Frau Geschlechtsverkehr haben.
Der verschreibende Arzt muss sicherstellen, dass männliche Patienten die Notwendigkeit der Verwendung eines Kondoms während der ganzen Behandlungsdauer, inklusive Behandlungsunterbrüchen und während 7 Tagen nach Abschluss der Behandlung verstehen und damit einverstanden sind, wenn sie mit einer schwangeren oder gebärfähigen Frau Geschlechtsverkehr haben.
Die Patienten dürfen während der Behandlung mit Imnovid und 7 Tage danach kein Sperma spenden.
Zusätzliche Vorsichtsmassnahmen
Die Patienten und Patientinnen müssen angewiesen werden, dieses Arzneimittel niemals anderen Personen zu geben und nicht verwendete Kapseln ihrem Arzt oder Apotheker nach Beendigung der Therapie zurückzugeben.
Informationsmaterial
- Informationsbroschüre für Patienten und Patientinnen zum Schwangerschaftsverhütungsprogramm von Pomalidomid mit Einverständniserklärung für Patienten und Patientinnen
- Vorgehen zur Schwangerschaftsverhütung
- Informationsbroschüre für Medizinalpersonen zum Schwangerschaftsverhütungsprogramm von Pomalidomid
Andere Warnhinweise und Vorsichtsmassnahmen
Unerwünschte hämatologische Wirkungen
Die Patienten sind auf hämatologische Toxizitäten, insbesondere Neutropenie, zu überwachen. Das vollständige Blutbild ist in den ersten 8 Wochen wöchentlich und danach monatlich zu kontrollieren. Eine Dosismodifikation kann erforderlich werden (siehe «Dosierung/Anwendung»).
Thromboembolische Ereignisse
Es wurde von venös thromboembolischen Ereignissen berichtet (vorwiegend tiefe Venenthrombosen und pulmonale Embolie). Deshalb wird eine Antikoagulation empfohlen (ausser wenn diese kontraindiziert ist). Die Entscheidung für Massnahmen zur Thrombose-Prophylaxe sollte nach Beurteilung der Risikofaktoren des jeweiligen Patienten mit der gebotenen Sorgfalt individuell getroffen werden.
Kardiale Erkrankungen
Fälle von Herzinsuffizienz, einschliesslich kongestive Herzinsuffizienz, Lungenödem und Vorhofflimmern wurden berichtet, vor allem bei Patienten mit vorbestehender Herzerkrankung oder kardialen Risikofaktoren. Wenn erwogen wird, solche Patienten mit Pomalidomid zu behandeln, ist entsprechende Vorsicht geboten, einschliesslich einer regelmässigen Überwachung auf Anzeichen und Symptome einer Herzinsuffizienz.
Tumorlyse-Syndrom
Es kann ein Tumorlyse-Syndrom auftreten. Bei Patienten mit einer hohen Tumorlast vor Behandlungsbeginn besteht das grösste Risiko für ein Tumorlyse-Syndrom. Diese Patienten sind engmaschig zu überwachen und es müssen geeignete Vorsichtsmassnahmen getroffen werden.
Allergische Reaktionen und schwere Hautreaktionen
Es wurde über Angioödeme, Anaphylaxie und schwere dermatologische Reaktionen, einschliesslich Stevens-Johnson-Syndrom (SJS), toxischer epidermaler Nekrolyse (TEN) und Arzneimittelexanthem mit Eosinophilie und systemischen Symptomen (DRESS), berichtet. Das DRESS-Syndrom kann sich in Form einer Hautreaktion (wie Hautausschlag oder exfoliative Dermatitis) in Verbindung mit Eosinophilie, Fieber und/oder Lymphadenopathie mit systemischen Komplikationen wie Hepatitis, Nephritis, Pneumonitis, Myokarditis und/oder Perikarditis zeigen. Diese Ereignisse können tödlich sein. Bei einem Grad-2- oder Grad-3-Hautausschlag sollte eine Unterbrechung oder das Absetzen der Behandlung mit Pomalidomid erwogen werden. Bei Angioödem, Anaphylaxie, Grad-4-Hautausschlag, exfoliativem oder bullösem Hautausschlag, oder bei Verdacht auf SJS, TEN oder DRESS muss Pomalidomid abgesetzt werden. Wurde die Behandlung wegen derartiger Reaktionen beendet, sollte sie nicht mehr aufgenommen werden.
Schwindel und Verwirrtheit
Es wurde über das Auftreten von Schwindel und Verwirrtheit berichtet. Weisen sie den Patienten zur Vorsicht auf Situationen hin, in denen Schwindel und Verwirrtheit ein Problem sein können.
Sekundäre Primärmalignome
Über sekundäre Primärmalignome wurde bei Patienten berichtet, die mit Pomalidomid behandelt wurden. Der Arzt/die Ärztin sollten die Patienten vor und während der Behandlung mithilfe der üblichen Massnahmen zur Krebsfrüherkennung hinsichtlich des Auftretens sekundärer Primärmalignome sorgfältig untersuchen und gegebenenfalls eine Therapie einleiten.
Leberfunktionsstörungen
Deutlich erhöhte Alaninaminotransferase- und Bilirubinspiegel wurden bei mit Pomalidomid behandelten Patienten beobachtet (siehe Abschnitt «Unerwünschte Wirkungen»). Es liegen auch Fälle von Hepatitis, einschliesslich Hepatitis B-Reaktivierung vor, die zum Absetzen von Pomalidomid führten. Eine regelmässige Kontrolle der Leberfunktion wird empfohlen.
Infektionen
Bei Patienten, die eine Kombinationstherapie mit Pomalidomid in klinischen Studien erhielten, traten bei 55,0-80,2% der Patienten Infektionen auf (24,0-30,9% Grad 3 oder 4). Infektionen der oberen Atemwege und Pneumonie waren die am häufigsten auftretenden Infektionen. Tödlich verlaufende Infektionen (Grad 5) traten bei 2,7-4,0% der Patienten auf. Infektionen führten bei 2,0-2,9% der Patienten zum Absetzen von Pomalidomid.
Patienten mit bekannten Risikofaktoren für das Auftreten von Infektionen müssen engmaschig überwacht werden. Alle Patienten sind anzuweisen, beim ersten Anzeichen einer Infektion (z.B. Husten, Fieber etc.) sofort einen Arzt aufzusuchen, um so durch eine frühzeitige Behandlung eine Verminderung des Schweregrades zu ermöglichen.
In seltenen Fällen wurde bei Patienten, die Pomalidomid in Kombination mit Dexamethason erhielten und zuvor mit dem Hepatitis-B-Virus (HBV) infiziert worden waren, über eine Reaktivierung von Hepatitis B berichtet. In einigen Fällen führte dies zu einem akuten Leberversagen, was ein Absetzen von Pomalidomid zur Folge hatte. Der Hepatitis-B-Virus-Status ist vor Beginn der Behandlung mit Pomalidomid abzuklären. Bei Patienten, die positiv auf eine HBV-Infektion getestet wurden, sollte ein Arzt mit Erfahrung in der Behandlung von Hepatitis B herangezogen werden. Entsprechende Vorsicht ist geboten, wenn Pomalidomid in Kombination mit Dexamethason bei vorher mit HBV infizierten Patienten angewendet wird. Diese Patienten müssen während der gesamten Behandlung engmaschig auf Anzeichen und Symptome einer aktiven HBV-Infektion überwacht werden.
Renale Störungen
Bei Patienten mit Kreatinin-Clearance ≤45 ml/min, die Pomalidomid in Kombination mit Bortezomib und Dexamethason erhielten, wurde eine erhöhte Rate an hämatologischen Nebenwirkungen (Anämie und Thrombozytopenie) und renalen Nebenwirkungen (akuter Nierenschaden) in klinischen Studien beobachtet (siehe auch «Eigenschaften/Wirkungen»). Solche Patienten sollten sorgfältig überwacht werden.
Zur Anwendung anderer Arzneimittel in Kombination mit Pomalidomid wird auf die entsprechende Fachinformation für das jeweilige Arzneimittel verwiesen.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Kapsel, d.h. es ist nahezu «natriumfrei».
Interaktionen
Potential anderer Arzneimittel zur Beeinflussung von Pomalidomid
Pomalidomid wird teilweise von CYP1A2 und CYP3A4/5 metabolisiert. Es ist ausserdem ein Substrat für P-Glycoprotein. Bei gleichzeitiger Anwendung von Pomalidomid mit Arzneistoffen, wie z.B. dem starken CYP3A4/5- und P-gp-Inhibitor Ketoconazol oder dem starken CYP3A4/5-Induktor Carbamazepin, wurde keine klinisch relevante Wirkung auf die Exposition gegenüber Pomalidomid festgestellt.
Die gleichzeitige Anwendung des starken CYP1A2-Hemmers Fluvoxamin mit Pomalidomid plus Ketoconazol erhöhte die durchschnittliche Exposition gegenüber Pomalidomid um 107% mit einem 90%-Konfidenzintervall [91% bis 124%] verglichen mit Pomalidomid plus Ketoconazol alleine. Die gleichzeitige Anwendung von Fluvoxamin (ein starker CYP1A2-Hemmer) mit Pomalidomid erhöhte die durchschnittliche Exposition gegenüber Pomalidomid um 125% mit einem 90%-Konfidenzintervall [98% bis 157%] verglichen mit Pomalidomid alleine bei gesunden Probanden. Wenn starke CYP1A2-Hemmer (z.B. Ciprofloxacin und Fluvoxamin) gleichzeitig mit Pomalidomid verabreicht werden, muss die Pomalidomid-Dosis um 50% reduziert werden.
Potential von Pomalidomid zur Beeinflussung anderer Arzneimittel
Pomalidomid hemmt nicht CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 oder CYP3A4/5 in vitro. Darüber hinaus ist Pomalidomid in vitro kein Induktor der Enzyme CYP1A2, CYP2B6, CYP2C9, CYP2C19 und CYP3A4/5.
Pomalidomid ist kein Inhibitor von P-Glycoprotein (P-gp) und zeigte in in-vitro-Untersuchungen keine oder eine nur geringe Hemmwirkung auf das Breast Cancer Resistant Protein (BCRP), Organic Anion Transporter Protein (OATP) 1B1, OATP1B3, Organic Cation Transporter OAT1 und OAT3 sowie Organic Anion Transporter Protein OCT2.
Es wird nicht damit gerechnet, dass Pomalidomid klinisch relevante pharmakokinetische Arzneimittel-Interaktionen infolge Enzymhemmung oder -induktion oder Transporter-Inhibition verursacht, wenn es zusammen mit Substraten dieser Enzyme oder Transporter angewendet wird. Das Potential für solche Arzneimittel-Interaktionen, und auch die potenziellen Auswirkungen von Pomalidomid auf die Exposition gegenüber oralen Kontrazeptiva, wurden klinisch nicht untersucht.
Schwangerschaft/Stillzeit
Schwangerschaft
Es ist mit einem teratogenen Effekt von Pomalidomid beim Menschen zu rechnen. Pomalidomid ist während der Schwangerschaft kontraindiziert und darf bei gebärfähigen Frauen nicht angewendet werden, ausser es sind alle Bedingungen für die Schwangerschaftsverhütung erfüllt (siehe «Warnhinweise und Vorsichtsmassnahmen»).
Stillzeit
Es ist nicht bekannt, ob Pomalidomid beim Menschen in die Muttermilch übergeht. Bei laktierenden Ratten wurde Pomalidomid nach Gabe an das Muttertier in der Milch wiedergefunden. Wegen seines Potenzials, bei gestillten Säuglingen Nebenwirkungen auszulösen, sollte unter Berücksichtigung der Bedeutung der Pomalidomid-Behandlung für die Mutter entschieden werden, ob abgestillt oder das Arzneimittel abgesetzt werden soll.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Es wurden keine Studien zu den Auswirkungen von Pomalidomid auf die Verkehrstüchtigkeit und das Bedienen von Maschinen durchgeführt. Unter der Anwendung von Pomalidomid wurde über Synkope, Fatigue und Schwindel berichtet. Daher wird zur Vorsicht geraten, wenn Patienten, welche Pomalidomid erhalten, am Verkehr teilnehmen oder Maschinen bedienen.
Unerwünschte Wirkungen
Imnovid in Kombination mit Bortezomib und Dexamethason (PVd) bei Patienten mit multiplem Myelom, die mindestens eine vorgängige Therapie erhalten haben
In der randomisierten Studie CC-4047-MM-007 erhielten 278 Patienten Pomalidomid, Bortezomib und Dexamethason.
Die am häufigsten gemeldeten Erkrankungen des Blutes und des Lymphsystems waren Neutropenie, Thrombozytopenie und Anämie. Die am häufigsten gemeldete unerwünschte Wirkung war periphere sensorische Neuropathie. Die am häufigsten gemeldete schwerwiegende unerwünschte Wirkung war Pneumonie (11,5%).
Imnovid in Kombination mit Dexamethason (Pd) bei Patienten mit rezidiviertem und refraktärem multiplem Myelom, welche mindestens zwei vorgängige Therapien erhalten haben
In drei klinischen Studien (CC-4047-MM-003, CC-4047-MM-002 und CC-4047-IFM-2009-02) wurden 455 Patienten gegenüber 4 mg Pomalidomid exponiert.
Die am häufigsten berichteten unerwünschten Wirkungen waren Neutropenie, Anämie, Thrombozytopenie, Fatigue, Fieber, Obstipation und Diarrhö.
Die häufigste schwerwiegende unerwünschte Wirkung, war Pneumonie (12,5%).
Die Häufigkeiten sind wie folgt definiert: Häufigkeitsangaben: sehr häufig (≥1/10); häufig (<1/10, ≥1/100); gelegentlich (<1/100, ≥1/1'000); selten (<1/1'000, ≥1/10'000), sehr selten (<1/10'000).
Infektionen und parasitäre Erkrankungen#
Sehr häufig: Infektionen der oberen Atemwege (20,9%), Pneumonie (19,1%), Bronchitis (14,0%), Virusinfektion der oberen Atemwege (11,2%).
Häufig: Sepsis, septischer Schock, Clostridium-difficile-Colitis, Bronchopneumonie, Influenza, Harnwegsinfektion, Atemwegsinfektion, Infektion der unteren Atemwege, Bronchiolitis, Lungeninfektion, Sinusitis, Nasopharyngitis, Candidiasis, orale Candidiasis.
Gelegentlich: Neutropenische Sepsis, Herpes Zoster*.
Selten: Reaktivierung des Hepatitis-B-Virus*.
# = Es werden alle bevorzugten Begriffe der Systemorganklasse Infektionen (einschliesslich bakterielle, virale und Pilzinfektionen), ausgenommen seltene Infektionen von öffentlichen Gesundheitsinteresse, aufgeführt.
Gutartige, bösartige und unspezifische Neubildungen (inklusive Zysten und Polypen)
Häufig: Basalzellkarzinom.
Gelegentlich: Plattenepithelkarzinom der Haut*.
Erkrankungen des Blutes und des Lymphsystems
Sehr häufig: Neutropenie (47,9%), Anämie (44,0%), Thrombozytopenie (36,7%), Leukopenie (13,0%).
Häufig: Febrile Neutropenie, Lymphopenie, verminderte Leukozytenzahl, verminderte Neutrophilenzahl, verminderte Thrombozytenzahl.
Gelegentlich: Panzytopenie*.
Erkrankungen des Immunsystem
Gelegentlich: Allergische Reaktionen (z.B. Angioödeme, Urtikaria)*.
Selten: Anaphylaxie*.
Endokrine Erkrankungen
Selten: Hypothyreose*.
Stoffwechsel- und Ernährungsstörungen
Sehr häufig: Hypokaliämie (15,5%), Hyperglykämie (14,4%), verminderter Appetit (11,6%).
Häufig: Dehydratation, Hyponatriämie, Hyperkaliämie, Hyperkalzämie, Hypokalzämie, Hypophosphatämie, Hypoalbuminämie, Hypomagnesiämie.
Psychiatrische Erkrankungen
Sehr häufig: Schlafstörungen (16,2%).
Häufig: Verwirrtheitszustand, Angstzustände, Depression, Stimmungsänderung.
Erkrankungen des Nervensystems
Sehr häufig: Periphere sensorische Neuropathie (47,8%), Schwindel (17,3%), Tremor (10,8%).
Häufig: Periphere Neuropathie, periphere sensorimotorische Neuropathie, Synkope, Somnolenz, Lethargie, Bewusstseinstrübung, Kopfschmerzen, Dysgeusie, Parästhesie.
Augenerkrankungen
Häufig: Verschwommenes Sehen, Katarakt.
Erkrankungen des Ohrs und Labyrinths
Häufig: Vertigo.
Herzerkrankungen
Häufig: Dekompensierte Herzinsuffizienz, Vorhofflimmern, Tachykardie.
Gefässerkrankungen
Häufig: Tiefe Venenthrombose, Hypotonie, Hypertonie.
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Sehr häufig: Husten (20,5%), Dyspnoe (20,2%).
Häufig: Lungenembolie, produktiver Husten, Nasenverstopfung, oropharyngeale Schmerzen, Dysphonie, Nasenbluten, Belastungsdyspnoe.
Gelegentlich: Atemnotsyndrom, interstitielle Lungenerkrankung einschliesslich Fälle mit Pneumonitis*.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Obstipation (36,7%), Diarrhö (33,8%), Übelkeit (17,6%), Erbrechen (11,5%).
Häufig: Bauchschmerzen, Oberbauchschmerzen, Stomatitis, trockener Mund, aufgeblähter Bauch.
Gelegentlich: Gastrointestinale Blutungen*.
Leber und Gallenerkrankungen
Häufig: Alaninaminotransferase erhöht.
Gelegentlich: Hyperbilirubinämie, erhöhte Leberfunktionswerte und Hepatitis*.
Erkrankungen der Haut und des Unterhautzellgewebes
Häufig: Hautausschlag, Pruritus, trockene Haut, Hyperhidrosis, Nachtschweiss.
Sehr selten: Stevens-Johnson-Syndrom*, toxische epidermale Nekrolyse*, Arzneimittelexanthem mit Eosinophilie und systemischen Symptomen*.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Sehr häufig: Rückenschmerzen (18,7%), Muskelschwäche (13,7%), Knochenschmerzen (13,6%), Muskelspasmen (13,0%).
Häufig: Arthralgie, Schmerzen des Bewegungsapparates, Schmerzen der Skelettmuskulatur im Brustbereich, Schmerzen in einer Extremität/Schmerzen in Gliedmassen.
Erkrankungen der Nieren und Harnwege
Häufig: Akuter Nierenschaden, Nierenversagen, akutes Nierenversagen, chronische Nierenerkrankung, Harnverhalt, Kreatininspiegel im Blut erhöht.
Erkrankungen der Geschlechtsorgane und der Brustdrüse
Häufig: Beckenschmerz.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Sehr häufig: Fatigue (37,1%), periphere Ödeme (33,8%), Fieber (23,7%), Asthenie (18,2%).
Häufig: Verschlechterung des körperlichen Allgemeinzustands, Schmerzen im Brustkorb, Schmerzen, Schüttelfrost, nicht herzbedingte Brustschmerzen, Ödem.
Gelegentlich: Tumorlyse-Syndrom*.
Untersuchungen
Häufig: Gewichtsverlust.
Verletzung, Vergiftung und durch Eingriffe bedingte Komplikationen
Häufig: Sturz.
* = Erfahrung nach Marktzulassung
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Pomalidomid-Dosen bis zu 50 mg als Einmalgabe bei gesunden Freiwilligen und 10 mg bei einmal täglicher wiederholter Verabreichung bei Patienten mit multiplem Myelom wurden untersucht, ohne dass überdosierungsbedingte schwerwiegende unerwünschte Ereignisse berichtet worden wären. Pomalidomid wurde durch Hämodialyse entfernt.
Eigenschaften/Wirkungen
ATC-Code
L04AX06
Wirkungsmechanismus
Pomalidomid ist ein Derivat von Thalidomid und liegt als Racemat vor. Es besitzt sowohl immunmodulierende als auch anti-angiogenetische Eigenschaften.
Pomalidomid hemmt die Proliferation hämatopoetischer Tumorzellen und hemmt in vitro auch die Proliferation Lenalidomid-resistenter Zelllinien. Es verstärkt die T-Zell Proliferation und die NK-Zell-vermittelte Zytotoxizität, hemmt die Produktion proinflammatorischer Zytokine (z.B. TNF-α und IL-6) und blockiert die Endothelzellmigration und -adhäsion.
Pomalidomid bindet direkt an das Protein Cereblon (CRBN), das Teil eines E3-Ligase-Komplexes ist, der DDB1-Protein (DNA damage-binding protein 1), Cullin 4 (CUL4) und Roc1 umfasst und die Autoubiquitinierung von CRBN innerhalb des Komplexes hemmen kann. E3-Ubiquitinligasen sind verantwortlich für die Polyubiquitinierung verschiedener Substratproteine und können teilweise die unter der Pomalidomid-Behandlung beobachteten pleiotropischen Zellwirkungen erklären.
In vitro, in Gegenwart von Pomalidomid, werden die Substratproteine Aiolos und Ikaros ubiquitiniert und anschliessend abgebaut, mit daraus resultierender direkter zytotoxischer und immunmodulatorischer Wirkung. In vivo führte die Pomalidomid-Therapie zur Abnahme des Ikaros-Spiegels bei Patienten mit rezidiviertem Lenalidomid-refraktärem multiplem Myelom.
Pharmakodynamik
Siehe Abschnitt Wirkungsmechanismus.
Klinische Wirksamkeit
Pomalidomid in Kombination mit Bortezomib und niedrig dosiertem Dexamethason (PVd)
Eine randomisierte, offene, 2-armige, multizentrische klinische Studie der Phase 3 (CC-4047-MM-007) bewertete die Wirksamkeit und Sicherheit von Pomalidomid in Kombination mit Bortezomib und niedrig dosiertem Dexamethason (PVd) im Vergleich zu Bortezomib und niedrig dosiertem Dexamethason (Vd) bei vorbehandelten erwachsenen Patienten, welche mindestens eine vorgängige Therapie erhalten haben, von denen eine Lenalidomid enthalten haben muss.
Die Patienten im PVd-Arm erhielten 4 mg Pomalidomid oral an den Tagen 1 bis 14 eines jeden 21-tägigen Zyklus. Niedrig dosiertes Dexamethason in einer Dosis von 20 mg wurde einmal täglich an den Tagen 1, 2, 4, 5, 8, 9, 11 und 12 eines 14-tägigen Zyklus in Zyklus 1 bis einschliesslich Zyklus 8 verabreicht und anschliessend einmal pro Tag an den Tagen 1, 2, 8 und 9 eines jeden anschliessenden 21-tägigen Zyklus von Zyklus 9 an. Patienten >75 Jahre erhielten Dexamethason 10 mg nach dem gleichen Therapieplan wie jüngere Patienten. Bortezomib (1,3 mg/m2/Dosis) wurde an den Tagen 1, 4, 8 und 11 des 21-tägigen Zyklus von Zyklus 1 bis einschliesslich Zyklus 8 verabreicht und anschliessend in der gleichen Dosis an den Tagen 1 und 8 des 21-tägigen Zyklus von Zyklus 9 an. Im Vd-Arm wurden Bortezomib und Dexamethason in der gleichen Dosis und nach dem gleichen Therapieplan verabreicht wie im PVd-Arm. Die Behandlung wurde bis zum Fortschreiten der Krankheit oder Auftreten einer nicht akzeptablen Toxizität fortgesetzt. Wenn es zur Behandlung von Toxizitätserscheinungen notwendig war, wurden die Dosen reduziert oder die Behandlung vorübergehend unterbrochen bzw. abgebrochen.
Es wurden insgesamt 559 Patienten in die Studie randomisiert: 281 in den PVd-Arm und 278 in den Vd-Arm. 54% der Patienten waren männlich bei einem medianen Alter der Gesamtpopulation von 68 Jahren (Min., Max.: 27, 89 Jahre). Ungefähr 70% der Patienten waren unter Lenalidomid refraktär (71,2% im PVd-Arm, 68,7% im Vd-Arm). Insgesamt stimmten die demographischen und krankheitsbezogenen Merkmale zwischen den Behandlungsarmen allgemein überein.
Der primäre Wirksamkeitsendpunkt war das progressionsfreie Überleben (PFS). Das PFS war definiert als die Zeit zwischen der Randomisierung und der Krankheitsprogression oder dem Tod. Das Ansprechen wurde von einem unabhängigen Ausschuss zur Beurteilung des Ansprechens (IRAC, Independent Response Adjudication Committee) entsprechend den IMWG-Kriterien unter Verwendung des Intent-to-Treat-Kollektivs als primäre Analyse beurteilt. Das Gesamtüberleben war ein sekundärer Endpunkt.
Beim ITT-Kollektiv, nach einer medianen Nachbeobachtung von 15,9 Monaten, betrug die Dauer des medianen PFS 11,20 Monate (95%-KI: 9,66; 13,73) im PVd-Arm. Im Vd-Arm betrug die Dauer des medianen PFS 7,10 Monate (95%-KI: 5,88; 8,48). Das PFS war im PVd-Arm signifikant länger als im Vd-Arm: HR 0,61 (95%-KI: 0,49; 0,77) und wies auf eine 39%ige Abnahme des Risikos für eine Krankheitsprogression oder für den Tod im PVd-Arm hin.
Bei Patienten mit Baseline CrCl <45 ml/min betrug das mediane PFS 7,16 Monaten (95% KI: 1,94; 10,74) im PVd-Arm (N = 37) verglichen mit 4,44 Monaten (95% KI: 2,83; 9,49) im Vd-Arm (N = 38); HR = 1,06, 95% KI: 0,61; 1,83.
Gemäss einer aktuellen Zwischenanalyse des Gesamtüberlebens (Daten Cut-off 15. September 2018; mediane Nachbeobachtung von 26,2 Monaten) waren 116/281 (41,3%) Patienten im PVd-Arm verstorben und 126/278 (45,3%) im Vd-Arm. Das mediane Gesamtüberleben aus der Kaplan-Meier-Schätzung betrug 40,5 Monate für den PVd-Arm und 30,5 Monate für den Vd-Arm; HR = 0,91, 95% KI: 0,70, 1,18, mit einer Gesamtereignis-Rate von 43,3%.
Pomalidomid in Kombination mit niedrig dosiertem Dexamethason (Pd)
Zur Beurteilung der Wirksamkeit und Sicherheit von Imnovid wurden eine Phase III Studie (CC-4047-MM-003) und eine Phase II Studie (CC-4047-MM-002) durchgeführt.
Die Phase II
Studie MM-002 wurde bei 221 Patienten mit rezidiviertem und refraktärem multiplem Myelom, welche gegenüber ihrer zuletzt erhaltenen Myelomtherapie refraktär waren und Lenalidomid und Bortezomib erhalten hatten, durchgeführt.
Pomalidomid 4 mg wurde bis zur Progredienz, entweder allein an 21/28 Tagen verabreicht oder in Kombination mit Dexamethason (40 mg wöchentlich, verabreicht an den Tagen 1, 8, 15 und 22 eines jeden 28-tägigen Zyklus).
Ca. 81% der Patienten waren gegenüber Lenalidomid, 74% gegenüber Bortezomib und 64% gegenüber Lenalidomid und Bortezomib refraktär (definiert nach den IMWG-Kriterien).
Das mediane progressionsfreie Überleben (Primärendpunkt) betrug bei Pomalidomid plus Dexamethason 16.6 Wochen und bei Pomalidomid allein 10,7 Wochen. Die Ansprechrate betrug 30,1% mit Pomalidomid plus Dexamethason gegenüber 9,3% mit Pomalidomid alleine.
Die Phase III
Die Phase III Studie (MM-003) verglich Pomalidomid plus Dexamethason-Therapie (Pom+LD-dex) mit Dexamethason allein (HD-dex) bei 455 Patienten mit rezidiviertem und refraktärem multiplem Myelom welche mindestens zwei Behandlungen erhalten hatten bei denen sowohl Lenalidomid als auch Bortezomib versagt hatten und die unter der letzten Therapie progredient waren.
Das mediane PFS (Primärendpunkt) betrug im POM+LD-dex Arm 15,7 Wochen (95% KI:13,0; 20,1) und 8 Wochen (95% KI: 7,0; 9,0) im HD-dex Arm, Hazard Ratio (HR) von 0,45 (95% KI: 0,35; 0,59; p <0,001).
Das mediane Gesamtüberleben wurde im Pomalidomid Arm noch nicht erreicht. Der Unterschied im Gesamtüberleben zwischen den beiden Behandlungsarmen war statistisch signifikant (Hazard Ratio von 0,53 (95% KI: 0,37; 0,74; p <0,001).
Pharmakokinetik
Absorption
Pomalidomid erreicht eine maximale Plasmakonzentration (Cmax) zwischen 2 und 3 Stunden p.a. und wird nach oraler Einmalgabe zu >70% resorbiert. Die Bioverfügbarkeit ist annähernd dosisproportional. Bei Gabe zusammen mit einer fett- und kalorienreichen Mahlzeit wird die Bioverfügbarkeit nicht beeinflusst. Pomalidomid kann unabhängig von den Mahlzeiten verabreicht werden.
Distribution
Das Verteilungsvolumen im Steady-State ist 62 bis 138 l. Die Konzentration in der Samenflüssigkeit gesunder Probanden betrug ca. 67% des Plasmaspiegels. Die Proteinbindung ist niedrig.
Metabolismus
Pomalidomid ist im zirkulierenden Blut die Hauptkomponente (etwa 70% der Plasmaradioaktivität). Es fanden sich keine Metaboliten, die >10% der Gesamtradioaktivität im Plasma ausmachten.
Die wichtigsten Stoffwechselwege der ausgeschiedenen Radioaktivität sind Hydroxylierung mit nachfolgender Glukuronidierung sowie Hydrolyse. In vitro wurden CYP1A2 und CYP3A4 als die an der CYP-vermittelten Hydroxylierung von Pomalidomid primär beteiligten Enzyme identifiziert, mit zusätzlichen geringfügigen Beiträgen von CYP2C19 und CYP2D6.
Bei 14 gesunden männlichen Probanden, die nach einer oralen Einzeldosis von 4 mg Pomalidomid über einen Zeitraum von 10 Tagen täglich 25 Zigaretten rauchten, kam es zu einem Anstieg der Cmax von Pomalidomid um 14%, während die AUC von Pomalidomid um 32% abnahm, im Vergleich zu 13 gesunden männlichen Freiwilligen, die Nichtraucher waren.
Elimination
Die Eliminationshalbwertszeit beträgt etwa 9,5 Stunden bei gesunden Probanden und etwa 7,5 Stunden bei Patienten mit multiplem Myelom. Die mittlere Gesamtkörper-Clearance (CL/F) beträgt 7-10 l/h.
73% der Dosis werden im Urin und 15% mit den Fäzes eliminiert, wobei als Pomalidomid etwa 2% bzw. 8% im Urin bzw. in den Fäzes ausgeschieden werden.
Kinetik spezieller Patientengruppen
Leberfunktionsstörungen
Die pharmakokinetischen Parameter waren bei Patienten mit eingeschränkter Leberfunktion (gemäss Child-Pugh-Kriterien) gering verändert verglichen mit gesunden Probanden. Die durchschnittliche Exposition gegenüber Pomalidomid war bei Patienten mit leicht eingeschränkter Leberfunktion verglichen mit gesunden Patienten um 51% erhöht bei einem 90%-Konfidenzintervall [9% bis 110%]. Die durchschnittliche Exposition gegenüber Pomalidomid war bei Patienten mit mässig eingeschränkter Leberfunktion im Vergleich zu gesunden Probanden um 58% erhöht bei einem 90%-Konfidenzintervall [13% bis 119%]. Die durchschnittliche Exposition gegenüber Pomalidomid war bei Patienten mit stark eingeschränkter Leberfunktion verglichen mit gesunden Patienten um 72% erhöht bei einem 90%-Konfidenzintervall [24% bis 138%].
Nierenfunktionsstörungen
Pharmakokinetische Populationsanalysen haben gezeigt, dass die pharmakokinetischen Parameter von Pomalidomid bei Patienten mit Nierenfunktionsstörung (definiert durch die Kreatinin-Clearance oder geschätzte glomeruläre Filtrationsrate [eGFR]) im Vergleich zu Patienten mit normaler Nierenfunktion (CrCl ≥60 ml/min) keine auffallende Beeinträchtigung aufwiesen. Die durchschnittliche normalisierte AUC-Exposition gegenüber Pomalidomid bei Patienten mit mässiger Niereninsuffizienz (eGFR ≥30 bis ≤45 ml/min/1.73 m2) betrug 98,2% mit einem 90%-Konfidenzintervall [77,4% bis 120,6%] im Vergleich zu Patienten mit normaler Nierenfunktion. Die durchschnittliche normalisierte AUC-Exposition gegenüber Pomalidomid bei nicht dialysepflichtigen Patienten mit schwerer Niereninsuffizienz (CrCl <30 oder eGFR <30 ml/min/1.73 m2) betrug 100,2% mit einem 90%-Konfidenzintervall [79,7% bis 127,0%] im Vergleich zu Patienten mit normaler Nierenfunktion. Die durchschnittliche normalisierte AUC-Exposition gegenüber Pomalidomid bei dialysepflichtigen Patienten mit schwerer Niereninsuffizienz (CrCl <30 ml/min bei Dialysepflicht) erhöhte sich um 35,8% mit einem 90%-Konfidenzintervall [7,5% bis 70,0%] im Vergleich zu Patienten mit normaler Nierenfunktion.
Ältere Patienten
Bei Patienten im Alter zwischen 61 und 82 Jahren waren die mittleren pharmakokinetischen Parameter von AUC (0-∞) und Cmax im Allgemeinen ähnlich zu denen jüngerer Patienten.
Kinder und Jugendliche
Daten zur Pharmakokinetik bei pädiatrischen Patienten liegen nicht vor.
Präklinische Daten
Genotoxizität/Karzinogenität
Pomalidomid war in Mutationstests an Bakterien und Säugerzellen nicht mutagen und induzierte in humanen peripheren Blutlymphozyten keine Chromosomenaberrationen und in polychromatischen Erythrozyten im Knochenmark von Ratten, denen Dosen bis zu 2'000 mg/kg/Tag verabreicht wurden, keine Mikrokernbildung. Karzinogenitätsstudien wurden nicht durchgeführt.
Fertilität und frühembryonale Entwicklung
In einer Studie bei Ratten zur Fertilität und frühembryonalen Entwicklung wurde Pomalidomid männlichen und weiblichen Tieren in Dosierungen von 25, 250 und 1'000 mg/kg/Tag verabreicht. Die Uterusuntersuchung am 13. Trächtigkeitstag ergab für alle Dosisstufen eine Abnahme der mittleren Anzahl überlebensfähiger Embryonen sowie vermehrte Postimplantationsverluste. Daher war das NOAEL für diese beobachteten Effekte <25 mg/kg/Tag (AUC24h – 39'960 ng·h/ml; 99-fach höhere Exposition bei der niedrigsten getesteten Dosis im Verhältnis zu einer 4-mg-Dosis). Wenn behandelte Männchen in dieser Studie mit unbehandelten Weibchen gepaart wurden, waren alle uterinen Parameter mit den Kontrollen vergleichbar. Auf der Grundlage dieser Resultate wurden die beobachteten Effekte der Behandlung der weiblichen Tiere zugeschrieben.
Embryo-fetale Entwicklung
Pomalidomid erwies sich sowohl bei Ratten als auch bei Kaninchen als teratogen, wenn es während der Phase der Hauptorganogenese verabreicht wurde. In der Studie zur embryo-fetalen Entwicklungstoxizität bei Ratten wurden in allen Dosisstufen (25, 250 und 1'000 mg/kg/Tag) Missbildungen oder Fehlen der Harnblase, Fehlen der Schilddrüse sowie Fusion und Fehlausrichtung lumbaler und thorakaler Wirbelelemente (Wirbelkörper- und/oder Neuralbögen) beobachtet. In dieser Studie wurde keine maternale Toxizität beobachtet. Daher betrug das maternale NOAEL 1'000 mg/kg/Tag und das NOAEL für Entwicklungstoxizität <25 mg/kg/Tag (AUC24h = 34'340 ng·h/ml am 17. Trächtigkeitstag; 85-fach höhere Exposition bei der niedrigsten getesteten Dosis im Verhältnis zur klinischen Dosis von 4 mg). Bei Kaninchen rief Pomalidomid in Dosierungen von 10 bis 250 mg/kg embryo-fetale Entwicklungsmissbildungen hervor. Bei allen Dosisstufen wurden vermehrt Herzanomalien beobachtet, wobei die Zunahme bei 250 mg/kg/Tag signifikant war. Bei 100 und 250 mg/kg/Tag fanden sich geringfügige Zunahmen der Postimplantationsverluste und geringfügige Abnahmen der fetalen Geburtsgewichte. Bei 250 mg/kg/Tag umfassten die fetalen Missbildungen Gliedmassenanomalien (geknickte und/oder rotierte Vorder- und/oder Hinterläufe, loser oder fehlender Digitus) und damit assoziierte Skelettfehlbildungen (nicht ossifiziertes Os metacarpale, Fehlausrichtung von Phalanx und Os metacarpale, fehlender Digitus, nicht ossifizierte Phalanx und kurze nicht ossifizierte oder geknickte Tibia); mässiggradige Dilatation des Lateralventrikels des Gehirns; Lageanomalie der A. subclavia dextra; fehlender Lungenmittellappen; tief liegende Niere; veränderte Lebermorphologie; unvollständig oder nicht ossifiziertes Becken; erhöhter Durchschnittswert überzähliger thorakaler Rippen und verminderter Durchschnittswert ossifizierter Ossa tarsalia. Eine geringfügige Verminderung der maternalen Körpergewichtszunahme, eine signifikante Reduktion der Triglyzeride und eine signifikante Abnahme der absoluten und relativen Milzgewichte wurden bei 100 und 250 mg/kg/Tag beobachtet. Das maternale NOAEL betrug 10 mg/kg/Tag, das NOAEL für die Entwicklung war <10 mg/kg/Tag (AUC24h = 418 ng·h/ml am 19. Trächtigkeitstag; 1-fache klinische Exposition bei der Dosis 4 mg).
Sonstige Hinweise
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
In der Originalverpackung, nicht über 25 °C und ausser Reichweite von Kindern aufbewahren.
Hinweise für die Handhabung
Wie bei den Zytostatika ist auch bei der Handhabung und Entsorgung von Imnovid besondere Vorsicht geboten (siehe auch «Dosierung/Anwendung»).
Gebärfähige Frauen, welche Pomalidomid handhaben, sollen Schutzhandschuhe tragen.
Die Kapseln dürfen weder geöffnet noch zerstossen werden. Falls Pomalidomid-Pulver mit der Haut in Kontakt kommt, ist die Haut sofort und gründlich mit Wasser und Seife zu waschen. Falls Pomalidomid mit Schleimhäuten in Kontakt kommt, ist gründlich mit Wasser zu spülen.
Zulassungsnummer
61249 (Swissmedic)
Zulassungsinhaberin
Celgene GmbH, Zürich
Stand der Information
Januar 2020
Composizione
Principi attivi
Pomalidomidum.
Sostanze ausiliarie
Mannitolo, amido pregelatinizzato, stearil fumarato di sodio.
Rivestimento della capsula: gelatina, biossido di titanio, ossido di ferro giallo (solo per capsule rigide 1 mg, 2 mg e 3 mg), indigocarmina E132, eritrosina E127 (solo per capsule rigide 2 mg), blu brillante FCF E133 (solo per capsule rigide 4 mg).
Inchiostro della dicitura: gommalacca, ossido di ferro nero, biossido di titanio, simeticone, glicole propilenico, soluzione di ammoniaca.
Una capsula rigida contiene max. 0,018 mg (capsule rigide 1 mg) o max. 0,036 mg (capsule rigide 2 mg) o max. 0,025 mg (capsule rigide 3 mg) o max. 0,033 (capsule rigide 4 mg) di sodio.
Forma farmaceutica e quantità di principio attivo per unità
Capsule rigide da 1 mg, 2 mg, 3 mg e 4 mg.
Indicazioni/Possibilità d'impiego
Imnovid, in associazione con bortezomib e desametasone, è indicato nel trattamento di pazienti adulti con mieloma multiplo (MM), sottoposti ad almeno una precedente terapia comprendente la lenalidomide.
Imnovid, in associazione con desametasone, è indicato nel trattamento di pazienti adulti con mieloma multiplo recidivato e refrattario, sottoposti ad almeno due precedenti terapie (comprendenti sia la lenalidomide che il bortezomib) e con dimostrata progressione della malattia durante l'ultima terapia.
Posologia/Impiego
Il trattamento deve essere iniziato e monitorato da ematologi od oncologi esperti.
Imnovid in associazione con bortezomib e desametasone (PVd) in pazienti con mieloma multiplo sottoposti ad almeno una precedente terapia
La dose iniziale raccomandata di Imnovid è di 4 mg una volta al giorno assunti per via orale nei giorni da 1 a 14 di cicli di trattamento ripetuti di 21 giorni.
La dose raccomandata di bortezomib è di 1,3 mg/m2 e la dose raccomandata di desametasone è di 20 mg/die per via orale una volta al giorno, secondo il regime posologico indicato nella Tabella 1.
Il dosaggio viene proseguito o modificato in base ai risultati clinici e di laboratorio. Il trattamento deve essere interrotto alla progressione della malattia.
Tabella 1. Regime posologico raccomandato per Imnovid in associazione con bortezomib e desametasone
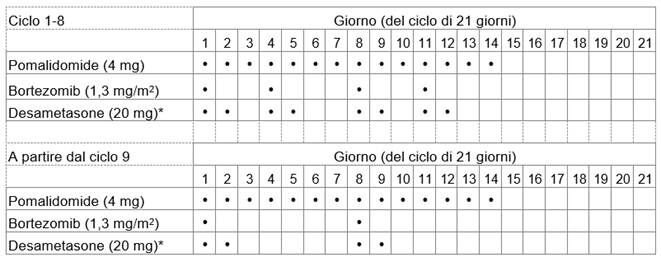
* Per i pazienti >75 anni di età, vedere paragrafo «Istruzioni posologiche speciali».
Imnovid in associazione con desametasone (Pd) in pazienti con mieloma multiplo recidivato e refrattario sottoposti ad almeno due precedenti terapie
La dose iniziale raccomandata di Imnovid è di 4 mg una volta al giorno assunti per via orale nei giorni da 1 a 21 di cicli di trattamento ripetuti di 28 giorni fino alla progressione della malattia. La dose raccomandata di desametasone è di 40 mg per via orale una volta al giorno nei giorni 1, 8, 15 e 22 di ogni ciclo di trattamento di trattamento di 28 giorni.
Il dosaggio viene proseguito o modificato in base ai risultati clinici e di laboratorio.
Il regime posologico è indicato nella Tabella 2.
Tabella 2. Regime posologico raccomandato per Imnovid in associazione con desametasone
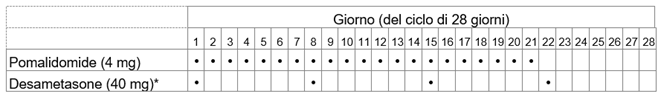
* Per i pazienti >75 anni di età, vedere paragrafo «Istruzioni posologiche speciali».
Aggiustamento della dose
Tossicità ematologica
In caso di trombocitopenia con calo dei valori a <25 x 109/l, o neutropenia con calo dei valori a <0,5 x 109/l, o neutropenia febbrile (febbre ≥38,5 °C e ANC <1,0 x 109/l), il trattamento con la pomalidomide deve essere interrotto, con successivi controlli settimanali del quadro emocromocitometrico (e in caso di calo della conta dei neutrofili, anche somministrazione di G-CSF a discrezione del medico curante). Dopo la normalizzazione della conta piastrinica/dei neutrofili, il trattamento con la pomalidomide deve essere continuato a una dose di 3 mg/die. Ad ogni successivo calo (rispettivamente <25 x 109/l e <0,5 x 109/l), il trattamento con la pomalidomide deve essere interrotto. Dopo la normalizzazione della conta dei trombociti/dei neutrofili, il trattamento con la pomalidomide deve essere ripreso a una dose ridotta di 1 mg rispetto all'ultima dose.
Imnovid in associazione con bortezomib e desametasone (PVd): per poter iniziare un nuovo ciclo di pomalidomide, la conta dei neutrofili deve essere ≥ 1 × 109/l e la conta piastrinica deve essere ≥50 × 109/l.
Imnovid in associazione con desametasone (Pd): per poter iniziare un nuovo ciclo di pomalidomide, la conta dei neutrofili deve essere ≥ 0,5 × 109/l e la conta piastrinica deve essere ≥ 50 × 109/l.
Altre tossicità di grado 3/4
Per le altre tossicità di grado 3/4 ritenute correlate alla pomalidomide, il trattamento deve essere interrotto e ripreso, a discrezione del medico, a una dose ridotta di 1 mg rispetto all'ultima dose, se la tossicità è regredita a ≤ grado 2. Se le rispettive tossicità si verificano anche dopo una riduzione della dose a 1 mg, la somministrazione del medicamento deve essere interrotta.
Per gli aggiustamenti della dose resi necessari dalla tossicità del bortezomib, consultare la relativa informazione professionale.
Aggiustamento della dose in caso di somministrazione concomitante di inibitori del CYP1A2
La somministrazione concomitante di pomalidomide e forti inibitori del CYP1A2 deve essere evitata. Considerare metodi di trattamento alternativi. In caso di somministrazione concomitante di pomalidomide e forti inibitori del CYP1A2 (ad es. ciprofloxacina e fluvoxamina), la dose di pomalidomide deve essere ridotta del 50%.
Interruzione del trattamento con la pomalidomide
In caso di eruzione cutanea di grado 2 o di grado 3, deve essere valutata la sospensione o l'interruzione del trattamento con la pomalidomide.
In caso di angioedema, anafilassi, eruzione cutanea di grado 4, eruzione cutanea esfoliativa o bollosa o in caso di sospetta sindrome di Stevens-Johnson (SJS), necrolisi epidermica tossica (TEN) o reazione da farmaco con eosinofilia e sintomi sistemici (DRESS), si deve interrompere il trattamento con la pomalidomide. Se il trattamento è stato interrotto per tali reazioni, non deve più essere ripreso.
Istruzioni posologiche speciali
Pazienti con disturbi della funzionalità epatica
Per i pazienti con lieve o moderata compromissione della funzionalità epatica (classi Child-Pugh A o B), la dose iniziale raccomandata è di 3 mg/die (riduzione della dose del 25%). Per i pazienti con compromissione della funzionalità epatica grave (classe Child-Pugh C), la dose iniziale raccomandata è di 2 mg/die (riduzione della dose del 50%).
Pazienti con disturbi della funzionalità renale
Nei pazienti con insufficienza renale moderata e nei pazienti con insufficienza renale grave che non richiedono dialisi non è necessario alcun aggiustamento della dose di pomalidomide. Nei pazienti dializzati con insufficienza renale grave, la dose iniziale raccomandata è 3 mg/die (riduzione della dose del 25%). Nei giorni in cui è prevista l'emodialisi, la pomalidomide deve essere assunta dopo la seduta di emodialisi.
Pazienti anziani
Non è necessario alcun aggiustamento della dose per la pomalidomide.
Imnovid in associazione con bortezomib e desametasone (PVd) dopo almeno una precedente terapia
Per i pazienti di età >75 anni, la dose di desametasone è di 10 mg una volta al giorno nei giorni 1, 2, 4, 5, 8, 9, 11 e 12 di un ciclo di 21 giorni, per i cicli da 1 a 8, e dal ciclo 9 in avanti è di 10 mg una volta al giorno nei giorni 1, 2, 8 e 9 di un ciclo di 21 giorni.
Imnovid in associazione con desametasone (Pd) dopo almeno 2 precedenti terapie
Per i pazienti di età >75 anni, la dose iniziale di desametasone è di 20 mg una volta al giorno nei giorni 1, 8, 15 e 22 di ogni ciclo di 28 giorni.
Bambini e adolescenti
Imnovid non è stata studiata nei pazienti pediatrici.
Modo di somministrazione
Imnovid deve essere assunta per via orale ogni giorno all'incirca alla stessa ora. Le capsule non devono essere aperte, spezzate o masticate. Le capsule di Imnovid devono essere deglutite senza essere masticate, preferibilmente con un po' d'acqua, con o senza assunzione di cibo. Se il paziente dimentica di assumere Imnovid un giorno, deve assumere la capsula successiva alla solita ora il giorno seguente. I pazienti non devono aumentare il numero di compresse da assumere per compensare la dose di Imnovid dimenticata il giorno precedente.
Per l'uso di altri medicamenti in associazione con la pomalidomide, consultare la relativa informazione professionale.
Controindicazioni
Ipersensibilità alla pomalidomide o a uno degli eccipienti, o alla talidomide e alla lenalidomide.
Gravidanza.
Donne potenzialmente fertili, a meno che non siano rispettate tutte le condizioni del Programma di prevenzione della gravidanza.
Per l'uso di altri medicamenti in associazione con la pomalidomide, consultare la relativa informazione professionale.
Avvertenze e misure precauzionali
La pomalidomide è un analogo della talidomide. La talidomide è una nota sostanza tossica per la riproduzione nell'uomo, che causa gravi difetti congeniti potenzialmente letali. La pomalidomide si è dimostrata tossica per la riproduzione sia nei ratti che nei conigli, quando è stata somministrata durante il periodo dell'organogenesi principale (vedere «Dati preclinici»). In caso di assunzione di pomalidomide durante la gravidanza è atteso un effetto teratogeno anche nell'uomo.
Programma di prevenzione della gravidanza
Programma per le pazienti di sesso femminile
Le condizioni del Programma di prevenzione della gravidanza devono essere soddisfatte per tutte le pazienti, a meno che non vi siano prove certe che la paziente non è in grado di concepire.
Criteri per stabilire che una donna non sia potenzialmente fertile
Una paziente di sesso femminile o la partner di un paziente di sesso maschile è considerata potenzialmente fertile a meno che non rispetti almeno uno dei seguenti criteri:
- Età ≥ 50 anni e amenorrea naturale per ≥ 1 anno*
- Insufficienza ovarica precoce confermata
- Pregressa salpingo-ovariectomia bilaterale, sterilizzazione tubarica o isterectomia
- Genotipo XY, sindrome di Turner, aplasia uterina
* L'amenorrea conseguente a una terapia antitumorale non esclude la potenziale fertilità.
Orientamento
La pomalidomide è controindicata per le donne potenzialmente fertili a meno che non siano soddisfatte tutte le seguenti condizioni:
- La paziente è consapevole del rischio teratogeno atteso per il feto.
- La paziente è consapevole della necessità di adottare metodi contraccettivi efficaci, senza interruzione, almeno 4 settimane prima di iniziare il trattamento, per l'intera durata del trattamento (inclusi i periodi di interruzione del trattamento) e fino a 4 settimane dopo la fine del trattamento.
- Anche in presenza di amenorrea, una paziente potenzialmente fertile deve seguire tutte le raccomandazioni per una contraccezione efficace.
- La paziente deve essere in grado di attenersi a misure contraccettive efficaci.
- La paziente è informata e consapevole delle conseguenze di una gravidanza e della necessità di consultare immediatamente il medico in caso di presunta gravidanza.
- La paziente è consapevole della necessità e accetta di sottoporsi a test di gravidanza ogni 4 settimane.
- La paziente dichiara di avere compreso i rischi e le precauzioni necessarie associate all'uso di pomalidomide.
In caso di donne potenzialmente fertili, il medico prescrivente deve assicurarsi che:
- La paziente soddisfi i criteri sopra indicati.
- La paziente rispetti le condizioni per la prevenzione della gravidanza, compresa la conferma che la paziente abbia un adeguato livello di comprensione.
- La paziente abbia utilizzato misure contraccettive sufficienti per almeno 4 settimane prima dell'inizio del trattamento, e che continui a utilizzare misure contraccettive efficaci per l'intera durata del trattamento (inclusi i periodi di interruzione del trattamento) e per almeno 4 settimane dopo la fine del trattamento. Le pazienti che necessitano di un trattamento immediato con la pomalidomide devono adottare una contraccezione adeguata, compreso l'uso di preservativi durante i 7 giorni precedenti l'inizio del trattamento.
- L'esito del test di gravidanza prima dell'inizio del trattamento sia negativo.
Contraccezione
Le donne potenzialmente fertili devono utilizzare metodi contraccettivi efficaci per 4 settimane prima dell'inizio del trattamento, per l'intera durata del trattamento (inclusi i periodi di interruzione del trattamento) e fino a 4 settimane dopo la fine del trattamento. Le pazienti che necessitano di un trattamento immediato con la pomalidomide devono adottare una contraccezione efficace, compreso l'uso di preservativi durante i 7 giorni precedenti l'inizio del trattamento. Nel caso in cui non sia stata già iniziata una terapia anticoncezionale efficace, la paziente deve essere indirizzata a un consultorio medico per ricevere una consulenza completa riguardo ai metodi contraccettivi efficaci.
Le procedure seguenti possono essere considerate metodi contraccettivi efficaci:
- Metodi indipendenti dalla paziente:
- Impianto
- Medrossiprogesterone acetato depot
- Sterilizzazione
- Metodi dipendenti dalla paziente:
- Astinenza dai rapporti sessuali eterosessuali
- Rapporti eterosessuali solo con partner di sesso maschile vasectomizzati; la vasectomia deve essere confermata da due analisi negative del liquido seminale
- Contraccettivi orali contenenti solo progesterone
A causa dell'aumentato rischio di tromboembolia venosa con la pomalidomide, è sconsigliato l'uso di contraccettivi orali di tipo combinato. Qualora la paziente utilizzi già contraccettivi orali di tipo combinato, deve considerare il passaggio a un diverso metodo contraccettivo. Il rischio di tromboembolia venosa permane per 4‑6 settimane dopo la sospensione del contraccettivo orale di tipo combinato. Qualora non sia possibile utilizzare altri metodi, si deve considerare una profilassi antitrombotica durante l'uso continuato dei contraccettivi orali di tipo combinato. La paziente deve essere adeguatamente informata in merito al rischio di tromboembolia venosa.
I sistemi intrauterini presentano un aumentato rischio di infezioni al momento dell'inserimento e possono causare sanguinamenti vaginali irregolari. Questi metodi non sono perciò raccomandati.
Test di gravidanza
Nelle pazienti potenzialmente fertili, si devono eseguire test di gravidanza con una sensibilità minima per l'hCG di 25 UI/ml.
Ogni caso di paziente con un test di gravidanza positivo deve essere immediatamente notificato allo Swiss Teratogen Information Service (STIS) di Losanna, utilizzando il formulario «Notifica di reazioni avverse (RA) da farmaci» di Swissmedic.
- Prima di iniziare il trattamento
Un test di gravidanza deve essere eseguito durante il consulto in cui viene prescritta la pomalidomide, oppure nei tre giorni precedenti la visita dal medico prescrivente, dopo che la paziente ha usato un metodo contraccettivo efficace per almeno 4 settimane. Il test deve garantire che la paziente non sia in stato di gravidanza al momento di iniziare il trattamento con la pomalidomide.
- Prima di iniziare il trattamento, se è necessario un trattamento immediato
Deve essere eseguito immediatamente un test quantitativo dell'hCG nel siero. Dopo una contraccezione efficace, compreso l'uso del preservativo per 7 giorni, questo test deve essere ripetuto. Qualora entrambi i test confermino che la paziente non è in gravidanza, è possibile iniziare il trattamento.
- Durante e al termine del trattamento
Un test di gravidanza deve essere ripetuto ogni 4 settimane e anche 4 settimane dopo la fine del trattamento. Questi test di gravidanza devono essere eseguiti durante la visita medica per la prescrizione della pomalidomide, oppure nei tre giorni precedenti.
Idealmente, i test di gravidanza, la prescrizione e la dispensazione della pomalidomide dovrebbero essere eseguiti nello stesso giorno. La dispensazione della pomalidomide deve essere effettuata entro un massimo di 7 giorni dopo la prescrizione.
Programma per i pazienti di sesso maschile
I dati preclinici dimostrano che nei pazienti di sesso maschile trattati con Imnovid questo principio attivo è presente nel liquido seminale. I pazienti di sesso maschile con partner potenzialmente fertili devono quindi usare il preservativo durante i rapporti sessuali, nel corso del trattamento con Imnovid e per almeno 7 giorni dopo il termine del trattamento. Gli uomini che assumono Imnovid devono soddisfare le seguenti condizioni:
- I pazienti devono essere consapevoli del rischio teratogeno atteso in caso di rapporto sessuale con una donna potenzialmente fertile.
- I pazienti devono essere consapevoli della necessità e accettare di utilizzare preservativi per l'intera la durata del trattamento (inclusi i periodi di interruzione del trattamento) e per 7 giorni dopo la fine del trattamento, nel caso in cui abbiano rapporti sessuali con una donna in stato di gravidanza o potenzialmente fertile.
Il medico prescrivente deve assicurarsi che i pazienti di sesso maschile siano consapevoli della necessità e accettino di utilizzare preservativi per l'intera la durata del trattamento (inclusi i periodi di interruzione del trattamento) e per 7 giorni dopo la fine del trattamento, nel caso in cui abbiano rapporti sessuali con una donna in stato di gravidanza o potenzialmente fertile.
I pazienti non devono donare liquido seminale durante il trattamento con Imnovid e per i 7 giorni successivi.
Ulteriori precauzioni di impiego
I pazienti devono essere istruiti a non dare mai questo medicamento ad altre persone e a restituire al medico o al farmacista le capsule non utilizzate dopo la fine del trattamento.
Materiale informativo
- Opuscolo informativo per i pazienti e le pazienti in merito al Programma di prevenzione della gravidanza per la pomalidomide, con modulo di consenso per i pazienti e le pazienti
- Misure di prevenzione della gravidanza
- Opuscolo informativo per gli operatori sanitari in merito al Programma di prevenzione della gravidanza per la pomalidomide
Altre avvertenze e misure precauzionali
Effetti indesiderati ematologici
I pazienti devono essere controllati per rilevare la comparsa di tossicità di natura ematologica, in particolare neutropenia. Il quadro emocromocitometrico deve essere monitorato settimanalmente per le prime 8 settimane e, successivamente, una volta al mese. Può essere necessaria una modifica della dose (vedere «Posologia / impiego»).
Eventi tromboembolici
Sono stati segnalati eventi tromboembolici venosi (principalmente trombosi venosa profonda ed embolia polmonare). Si raccomanda perciò una terapia anticoagulante (salvo controindicazioni). La decisione di adottare misure profilattiche antitrombotiche deve essere presa dopo un'attenta valutazione dei fattori di rischio del singolo paziente.
Patologie cardiache
Sono stati segnalati casi di insufficienza cardiaca, inclusa insufficienza cardiaca congestizia, edema polmonare e fibrillazione atriale, soprattutto in pazienti con cardiopatia preesistente o fattori di rischio cardiaco. Nel considerare il trattamento di tali pazienti con la pomalidomide occorre usare adeguata cautela, incluso il monitoraggio periodico per rilevare eventuali segni o sintomi di insufficienza cardiaca.
Sindrome da lisi tumorale
Può verificarsi sindrome da lisi tumorale. I pazienti a maggior rischio di sindrome da lisi tumorale sono quelli con carico tumorale elevato prima del trattamento. Si raccomanda di monitorare attentamente tali pazienti e di adottare le precauzioni appropriate.
Reazioni allergiche e reazioni cutanee gravi
Sono stati riportati casi di angioedema, anafilassi e reazioni dermatologiche gravi, incluse sindrome di Stevens-Johnson (SJS), necrolisi epidermica tossica (TEN) e reazione da farmaco con eosinofilia e sintomi sistemici (DRESS). La sindrome DRESS può manifestarsi sotto forma di reazione cutanea (come eruzione cutanea o dermatite esfoliativa) associata a eosinofilia, febbre e/o linfoadenopatia con complicanze sistemiche quali epatite, nefrite, polmonite, miocardite e/o pericardite. Questi eventi possono avere esito fatale. In caso di eruzione cutanea di grado 2 o di grado 3, deve essere valutata la sospensione o l'interruzione del trattamento con la pomalidomide. In caso di angioedema, anafilassi, eruzione cutanea di grado 4, eruzione cutanea esfoliativa o bollosa o in caso di sospetta SJS, TEN o DRESS si deve interrompere il trattamento con la pomalidomide. Se il trattamento è stato interrotto per reazioni di tale natura, non deve più essere ripreso.
Capogiri e confusione
È stata segnalata la comparsa di capogiri e stato confusionale. I pazienti devono essere avvertiti di usare cautela nelle situazioni in cui capogiri o confusione possono rappresentare un problema.
Secondi tumori maligni primari
Sono stati segnalati secondi tumori maligni primari in pazienti trattati con la pomalidomide. Il medico deve valutare attentamente i pazienti, prima e durante il trattamento, mediante le misure di screening oncologico standard, relativamente alla comparsa di secondi tumori maligni primari ed eventualmente istituire una terapia.
Disturbi della funzionalità epatica
Nei pazienti trattati con la pomalidomide sono stati osservati livelli ematici marcatamente elevati di alanina aminotransferasi e bilirubina (vedere paragrafo «Effetti indesiderati»). Vi sono stati inoltre casi di epatite, inclusa riattivazione dell'epatite B, che hanno comportato l'interruzione del trattamento con la pomalidomide. Si raccomanda un monitoraggio regolare della funzionalità epatica.
Infezioni
Nei pazienti sottoposti a una terapia combinata con la pomalidomide negli studi clinici, si sono manifestate infezioni nel 55,0‑80,2% dei pazienti (24,0-30,9% di grado 3 o 4). Infezioni delle vie respiratorie superiori e polmonite sono state le infezioni più comunemente osservate. Infezioni con esito fatale (grado 5) si sono verificate nel 2,7‑4,0% dei pazienti. Nel 2,0‑2,9% dei pazienti le infezioni hanno comportato la sospensione del trattamento con la pomalidomide.
I pazienti con fattori di rischio noti per la comparsa di infezioni devono essere attentamente monitorati. Tutti i pazienti devono essere avvisati di consultare immediatamente un medico al primo segno di infezione (es. tosse, febbre, ecc.) al fine di ridurne la gravità attraverso il trattamento precoce.
In rari casi, nei pazienti trattati con pomalidomide in associazione a desametasone, con pregressa infezione da virus dell'epatite B (HBV), è stata segnalata la riattivazione dell'epatite B. Alcuni di questi casi sono progrediti in insufficienza epatica acuta, che ha comportato l'interruzione del trattamento con la pomalidomide. Lo stato virale dell'epatite B deve essere stabilito prima di iniziare il trattamento con la pomalidomide. Per i pazienti che risultano positivi al test per l'infezione da HBV, si raccomanda di consultare un medico esperto nel trattamento dell'epatite B. Si deve usare cautela quando si utilizza la pomalidomide in associazione al desametasone in pazienti con pregressa infezione da HBV. Questi pazienti devono essere attentamente monitorati per rilevare segni e sintomi di infezione da HBV attiva durante la terapia.
Disturbi renali
Nei pazienti con clearance della creatinina ≤45 ml/min trattati negli studi clinici con la pomalidomide in associazione con bortezomib e desametasone è stato osservato un aumento del tasso di effetti indesiderati ematologici (anemia e trombocitopenia) e di effetti indesiderati renali (danno renale acuto) (vedere anche «Proprietà / effetti»). Tali pazienti devono essere attentamente monitorati.
Per l'uso di altri medicamenti in associazione con la pomalidomide, consultare la relativa informazione professionale.
Questo medicamento contiene meno di 1 mmol di sodio (23 mg) per capsula, ovvero è quasi «privo di sodio».
Interazioni
Potenziale effetto di altri medicamenti sulla pomalidomide
La pomalidomide è metabolizzata in parte dal CYP1A2 e dal CYP3A4/5. È inoltre un substrato per la glicoproteina-P. La somministrazione concomitante di pomalidomide e medicamenti quali ketoconazolo, un forte inibitore del CYP3A4/5 e della P-gp, o carbamazepina, un forte induttore del CYP3A4/5, non ha avuto un effetto clinicamente rilevante sull'esposizione alla pomalidomide.
La somministrazione concomitante di fluvoxamina, un forte inibitore del CYP1A2, e pomalidomide più ketoconazolo ha aumentato l'esposizione media alla pomalidomide del 107% (intervallo di confidenza al 90%: da 91% a 124%) rispetto a pomalidomide più ketoconazolo da soli. In soggetti sani, la somministrazione concomitante di fluvoxamina (un forte inibitore del CYP1A2) e pomalidomide ha aumentato l'esposizione media alla pomalidomide del 125% (intervallo di confidenza al 90%: da 98% a 157%) rispetto alla pomalidomide da sola. In caso di somministrazione concomitante di pomalidomide e forti inibitori del CYP1A2 (ad es. ciprofloxacina e fluvoxamina), la dose di pomalidomide deve essere ridotta del 50%.
Potenziale effetto della pomalidomide su altri medicamenti
La pomalidomide non inibisce CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 o CYP3A4/5 in vitro. Inoltre, in vitro la pomalidomide non è risultata un induttore degli enzimi CYP1A2, CYP2B6, CYP2C9, CYP2C19 e CYP3A4/5.
La pomalidomide non è un inibitore della glicoproteina-P (P‑gp) e negli studi in vitro non ha evidenziato alcun effetto inibitore, o ha evidenziato un effetto inibitore minimo, su Breast Cancer Resistant Protein (BCRP), Organic Anion Transporter Protein (OATP) 1B1, OATP1B3, Organic Cation Transporter OAT1 e OAT3 nonché Organic Anion Transporter Protein OCT2.
Non si prevede che la pomalidomide causi interazioni farmacocinetiche farmaco-farmaco di rilievo clinico, in seguito a inibizione o induzione enzimatica o a inibizione dei trasportatori, nel caso in cui sia somministrata in concomitanza con substrati di tali enzimi o trasportatori. Il potenziale di tali interazioni farmacologiche, inclusi gli effetti potenziali della pomalidomide sull'esposizione ai contraccettivi orali, non è stato valutato dal punto di vista clinico.
Gravidanza/Allattamento
Gravidanza
È atteso un effetto teratogeno della pomalidomide nell'uomo. La pomalidomide è controindicata durante la gravidanza e non deve essere usata nelle donne potenzialmente fertili a meno che non siano soddisfatte tutte le condizioni per la prevenzione della gravidanza (vedere «Avvertenze e misure precauzionali»).
Allattamento
Non è noto se la pomalidomide sia escreta nel latte materno umano. La pomalidomide è stata rilevata nel latte di ratti in lattazione dopo somministrazione alla madre. Dato il potenziale della pomalidomide di causare effetti indesiderati nei neonati allattati con latte materno, si deve decidere se sospendere l'allattamento o l'assunzione del medicamento, tenendo conto dell'importanza della terapia per la madre.
Effetti sulla capacità di condurre veicoli e sull'impiego di macchine
Non sono stati effettuati studi in merito agli effetti della pomalidomide sulla capacità di condurre veicoli e sull'impiego di macchine. Con l'uso di pomalidomide sono stati segnalati sincope, affaticamento e capogiro. Pertanto, ai pazienti trattati con la pomalidomide si consiglia cautela nella partecipazione al traffico stradale e nell'impiego di macchine.
Effetti indesiderati
Imnovid in associazione con bortezomib e desametasone (PVd) in pazienti con mieloma multiplo sottoposti ad almeno una precedente terapia
Nello studio randomizzato CC-4047-MM-007, 278 pazienti hanno ricevuto pomalidomide, bortezomib e desametasone.
Le patologie del sistema emolinfopoietico più comunemente segnalate sono state neutropenia, trombocitopenia e anemia. L'effetto indesiderato più comunemente segnalato è stata la neuropatia sensitiva periferica. L'effetto indesiderato grave più comunemente segnalato è stato la polmonite (11,5%).
Imnovid in associazione con desametasone (Pd) in pazienti con mieloma multiplo recidivato e refrattario sottoposti ad almeno due precedenti terapie
In tre studi clinici (CC-4047-MM-003, CC-4047-MM-002 e CC-4047-IFM-2009-02), 455 pazienti sono stati esposti alla pomalidomide a una dose di 4 mg.
Gli effetti indesiderati più comunemente segnalati sono stati neutropenia, anemia, trombocitopenia, affaticamento, febbre, stipsi e diarrea.
L'effetto indesiderato grave più comune è stata la polmonite (12,5%).
Le frequenze sono definite come segue: molto comune (≥1/10); comune (<1/10, ≥1/100); non comune (<1/100, ≥1/1000); raro (<1/1000, ≥1/10'000), molto raro (<1/10'000).
Infezioni ed infestazioni#
Molto comune: Infezioni delle vie respiratorie superiori (20,9%), polmonite (19,1%), bronchite (14,0%), infezione virale delle vie respiratorie superiori (11,2%).
Comune: Sepsi, shock settico, colite da Clostridium difficile, broncopolmonite, influenza, infezione delle vie urinarie, infezione delle vie respiratorie, infezione delle vie respiratorie inferiori, bronchiolite, infezione polmonare, sinusite, nasofaringite, candidosi, candidosi orale.
Non comune: Sepsi neutropenica, herpes zoster*.
Raro: Riattivazione del virus dell'epatite B*.
# = Sono elencati tutti i termini preferiti della classe sistemica-organica «Infezioni» (comprese le infezioni batteriche, virali e micotiche), ad eccezione delle infezioni rare di interesse sanitario pubblico.
Tumori benigni, maligni e non specificati (cisti e polipi compresi)
Comune: Carcinoma basocellulare.
Non comune: Carcinoma cutaneo squamocellulare*.
Patologie del sistema emolinfopoietico
Molto comune: Neutropenia (47,9%), anemia (44,0%), trombocitopenia (36,7%), leucopenia (13,0%).
Comune: Neutropenia febbrile, linfopenia, riduzione della conta leucocitaria, riduzione della conta dei neutrofili, riduzione della conta piastrinica.
Non comune: Pancitopenia*.
Disturbi del sistema immunitario
Non comune: Reazioni allergiche (ad es. angioedema, orticaria)*.
Raro: Anafilassi*.
Patologie endocrine
Raro: Ipotiroidismo*.
Disturbi del metabolismo e della nutrizione
Molto comune: Ipokaliemia (15,5%), iperglicemia (14,4%), riduzione dell'appetito (11,6%).
Comune: Disidratazione, iponatriemia, ipokaliemia, ipercalcemia, ipocalcemia, ipofosfatemia, ipoalbuminemia, ipomagnesiemia.
Disturbi psichiatrici
Molto comune: Disturbi del sonno (16,2%).
Comune: Stato confusionale, ansia, depressione, alterazione dell'umore.
Patologie del sistema nervoso
Molto comune: Neuropatia sensitiva periferica (47,8%), capogiro (17,3%), tremore (10,8%).
Comune: Neuropatia periferica, neuropatia sensitivo-motoria periferica, sincope, sonnolenza, letargia, obnubilazione, cefalea, disgeusia, parestesia.
Patologie dell'occhio
Comune: Annebbiamento della vista, cataratta.
Patologie dell'orecchio e del labirinto
Comune: Vertigine.
Patologie cardiache
Comune: Insufficienza cardiaca scompensata, fibrillazione atriale, tachicardia.
Patologie vascolari
Comune: Trombosi venosa profonda, ipotensione, ipertensione.
Patologie respiratorie, toraciche e mediastiniche
Molto comune: Tosse (20,5%), dispnea (20,2%).
Comune: Embolia polmonare, tosse produttiva, congestione nasale, dolore orofaringeo, disfonia, epistassi, dispnea da sforzo.
Non comune: Sindrome da sofferenza respiratoria, malattia polmonare interstiziale inclusi casi con polmonite*.
Patologie gastrointestinali
Molto comune: Stipsi (36,7%), diarrea (33,8%), nausea (17,6%), vomito (11,5%).
Comune: Dolore addominale, dolore epigastrico, stomatite, secchezza della bocca, distensione addominale.
Non comune: Emorragie gastrointestinali*.
Patologie epatobiliari
Comune: Alanina aminotransferasi aumentata.
Non comune: Iperbilirubinemia, aumento dei valori di funzionalità epatica ed epatite*.
Patologie della cute e del tessuto sottocutaneo
Comune: Eruzione cutanea, prurito, secchezza cutanea, iperidrosi, sudorazione notturna.
Molto raro: Sindrome di Stevens-Johnson*, necrolisi epidermica tossica*, reazione da farmaci con eosinofilia e sintomi sistemici*.
Patologie del sistema muscoloscheletrico e del tessuto connettivo
Molto comune: Dolore dorsale (18,7%), debolezza muscolare (13,7%), dolore osseo (13,6%), spasmi muscolari (13,0%).
Comune: Artralgia, dolore all'apparato locomotore, dolore alla muscolatura scheletrica nella zona toracica, dolore a un arto/agli arti.
Patologie renali e urinarie
Comune: Danno renale acuto, insufficienza renale, insufficienza renale acuta, patologia renale cronica, ritenzione urinaria, aumento della creatininemia.
Patologie dell'apparato riproduttivo e della mammella
Comune: Dolore pelvico.
Patologie sistemiche e condizioni relative alla sede di somministrazione
Molto comune: Affaticamento (37,1%), edema periferico (33,8%), febbre (23,7%), astenia (18,2%).
Comune: Deterioramento dello stato di salute fisica generale, dolore toracico, dolore, brividi, dolore toracico non cardiaco, edema.
Non comune: Sindrome da lisi tumorale*.
Esami diagnostici
Comune: Perdita di peso.
Traumatismo, avvelenamento e complicazioni da procedura
Comune: Caduta.
* = esperienza post-marketing
La notifica di effetti collaterali sospetti dopo l'omologazione del medicamento è molto importante. Consente una sorveglianza continua del rapporto rischio-benefico del medicamento. Chi esercita una professione sanitaria è invitato a segnalare qualsiasi nuovo o grave effetto collaterale sospetto attraverso il portale online ElViS (Electronic Vigilance System). Maggiori informazioni sul sito www.swissmedic.ch.
Posologia eccessiva
Sono state studiate dosi di pomalidomide fino a 50 mg, come dose singola in volontari sani, e fino a 10 mg una volta al giorno come somministrazione ripetuta in pazienti con mieloma multiplo, senza segnalazione di eventi avversi seri correlati a sovradosaggio. La pomalidomide è stata eliminata mediante emodialisi.
Proprietà/Effetti
Codice ATC
L04AX06
Meccanismo d'azione
La pomalidomide è un derivato della talidomide ed è presente come racemato. Possiede proprietà sia immunomodulatorie sia antiangiogeniche.
La pomalidomide inibisce la proliferazione delle cellule tumorali ematopoietiche e inibisce in vitro anche la proliferazione delle linee cellulari resistenti alla lenalidomide. La pomalidomide potenzia la proliferazione dei linfociti T e la citotossicità mediata dalle cellule NK, inibisce la produzione di citochine proinfiammatorie (ad es. TNF-α e IL 6) e blocca la migrazione e l'adesione delle cellule endoteliali. La pomalidomide si lega direttamente alla proteina cereblon (CRBN), che fa parte di un complesso E3 ligasi comprendente la proteina DDB1 (DNA Damage-Binding Protein 1), cullina 4 (CUL4) e Roc1, ed è in grado di inibire l'auto-ubiquitinazione della CRBN all'interno del complesso. Le ubiquitina E3 ligasi sono responsabili della poli-ubiquitinazione di svariate proteine substrato e possono in parte spiegare gli effetti cellulari pleiotropici osservati nel trattamento con la pomalidomide.
In vitro, in presenza di pomalidomide, le proteine substrato Aiolos e Ikaros sono oggetto di ubiquitinazione e successiva degradazione, con conseguenti effetti citotossici e immunomodulatori diretti. In vivo, la terapia con la pomalidomide ha prodotto una riduzione dei livelli di Ikaros nei pazienti affetti da mieloma multiplo recidivato refrattario alla lenalidomide.
Farmacodinamica
Vedere la sezione sul meccanismo d'azione.
Efficacia clinica
Pomalidomide in associazione con bortezomib e desametasone a basso dosaggio (PVd)
L'efficacia e la sicurezza della pomalidomide in associazione con bortezomib e desametasone a basso dosaggio (PVd) sono state valutate, rispetto a bortezomib e desametasone a basso dosaggio (Vd), in uno studio clinico di fase 3 multicentrico, randomizzato, in aperto, a 2 bracci (CC-4047-MM-007), condotto in pazienti adulti sottoposti ad almeno una precedente terapia comprendente la lenalidomide.
Ai pazienti del braccio PVd sono stati somministrati 4 mg di pomalidomide per via orale nei giorni da 1 a 14 di ogni ciclo di 21 giorni. Il desametasone a basso dosaggio, a una dose di 20 mg, è stato somministrato, una volta al giorno, i giorni 1, 2, 4, 5, 8, 9, 11 e 12 di un ciclo di 14 giorni, per i cicli da 1 a 8 compreso; successivamente una volta al giorno i giorni 1, 2, 8 e 9 di ciascun ciclo successivo di 21 giorni, per i cicli da 9 in poi. I pazienti >75 anni di età hanno ricevuto 10 mg di desametasone secondo lo stesso piano terapeutico previsto per i pazienti più giovani. Il bortezomib (1,3 mg/m2/dose) è stato somministrato i giorni 1, 4, 8 e 11 di un ciclo di 21 giorni, per i cicli da 1 a 8 compreso e successivamente, alla stessa dose, i giorni 1 e 8 di ciascun ciclo successivo di 21 giorni per i cicli da 9 in poi. Nel braccio Vd, il bortezomib e il desametasone sono stati somministrati alla stessa dose e secondo lo stesso piano terapeutico del braccio PVd. Il trattamento è stato continuato fino alla progressione della malattia o al verificarsi di una tossicità inaccettabile. Se necessario per gestire le manifestazioni di tossicità, le dosi sono state ridotte oppure il trattamento è stato temporaneamente sospeso o interrotto.
Nello studio sono stati randomizzati in totale 559 pazienti: 281 nel braccio PVd e 278 nel braccio Vd. Il 54% dei pazienti era di sesso maschile, con un'età mediana della popolazione complessiva di 68 anni (min 27 anni, max 89 anni). Circa il 70% dei pazienti era refrattario alla lenalidomide (71,2% nel braccio PVd, 68,7% nel braccio Vd). Nel complesso, le caratteristiche demografiche e quelle correlate alla malattia dei diversi bracci di trattamento sono state generalmente sovrapponibili.
L'endpoint primario di efficacia è stata la sopravvivenza libera da progressione (PFS), definita come il tempo intercorrente tra la randomizzazione e la progressione della malattia o il decesso. La risposta è stata valutata da un comitato indipendente per la valutazione della risposta (Independent Response Adjudication Committee, IRAC) secondo i criteri del gruppo internazionale di lavoro sul mieloma (IMWG) utilizzando la popolazione intention-to-treat (ITT) come analisi primaria. L'endpoint secondario è stata la sopravvivenza globale.
Nella popolazione ITT, dopo un follow-up mediano di 15,9 mesi, il tempo mediano di PFS è stato di 11,20 mesi (IC al 95%: 9,66; 13,73) nel braccio PVd. Nel braccio Vd, il tempo mediano di PFS è stato di 7,10 mesi (IC al 95%: 5,88; 8,48). La PFS è risultata significativamente più lunga nel braccio PVd rispetto al braccio Vd: HR 0,61 (IC al 95%: 0,49; 0,77) e ha evidenziato una riduzione del 39% del rischio di progressione della malattia o di decesso nel braccio PVd.
Nei pazienti con CrCl al basale <45 ml/min, la PFS mediana è stata pari a 7,16 mesi (IC al 95%: 1,94, 10,74) nel braccio PVd (n = 37), rispetto a 4,44 mesi (IC al 95%: 2,83, 9,49) nel braccio Vd (n = 38); HR = 1,06 (IC al 95%: 0,61; 1,83).
Secondo un'analisi ad interim aggiornata per la sopravvivenza globale (cut-off dei dati al 15 settembre 2018, follow-up mediano di 26,2 mesi), erano deceduti 116/281 (41,3%) pazienti nel braccio PVd e 126/278 (45,3%) pazienti nel braccio Vd. La sopravvivenza globale mediana secondo le stime di Kaplan-Meier è stata di 40,5 mesi per il braccio PVd e di 30,5 mesi per il braccio Vd; HR = 0,91 (IC al 95%: 0,70, 1,18) con un tasso globale di eventi del 43,3%.
Pomalidomide in associazione con desametasone a basso dosaggio (Pd)
L'efficacia e la sicurezza di Imnovid sono state valutate in uno studio di fase III (CC-4047-MM-003) e in uno studio di fase II (CC-4047-MM-002).
Fase II
Lo studio di fase II MM-002 è stato condotto in 221 pazienti affetti da mieloma multiplo recidivato e refrattario, che erano risultati refrattari all'ultima terapia per il mieloma ricevuta e che erano stati trattati con lenalidomide e bortezomib.
La pomalidomide alla dose di 4 mg è stata somministrata fino alla progressione della malattia in monoterapia (con cicli di 21/28 giorni) o in associazione con il desametasone (40 mg alla settimana, somministrati i giorni 1, 8, 15 e 22 di ogni ciclo di 28 giorni).
Circa l'81% dei pazienti era refrattario alla lenalidomide, il 74% al bortezomib e il 64% alla lenalidomide e al bortezomib (definiti secondo i criteri IMWG).
Il valore mediano della sopravvivenza libera da progressione (endpoint primario) è stato pari a 16,6 settimane con pomalidomide più desametasone, e a 10,7 settimane con pomalidomide in monoterapia. Il tasso di risposta è stato del 30,1% con pomalidomide più desametasone, rispetto al 9,3% con pomalidomide in monoterapia.
Fase III
Lo studio di fase III (MM-003) ha confrontato la terapia con pomalidomide più desametasone (Pom +LD Dex) con desametasone in monoterapia (HD-Dex) in 455 pazienti affetti da mieloma multiplo recidivato e refrattario, sottoposti ad almeno due precedenti trattamenti nei quali sia la lenalidomide che il bortezomib erano risultati inefficaci e che avevano evidenziato una progressione della malattia durante l'ultima terapia.
La PFS mediana (endpoint primario) nel braccio POM+LD-dex è stata di 15,7 settimane (IC al 95%: 13,0; 20,1) e di 8 settimane (IC al 95%: 7,0; 9,0) nel braccio HD-dex, hazard ratio (HR) di 0,45 (IC al 95%: 0,35; 0,59; p <0,001).
La sopravvivenza globale mediana nel braccio pomalidomide non è stata ancora raggiunta. La differenza in termini di sopravvivenza globale tra i due bracci di trattamento è stata statisticamente significativa (hazard ratio di 0,53 [IC al 95%: 0,37; 0,74]; p <0,001).
Farmacocinetica
Assorbimento
La pomalidomide raggiunge una concentrazione plasmatica massima (Cmax) in un tempo compreso tra 2 e 3 ore dopo la somministrazione e viene assorbita per >70% dopo la somministrazione di una dose orale singola. La biodisponibilità è quasi proporzionale alla dose. La somministrazione insieme a un pasto ipercalorico e ad alto contenuto di grassi non influisce sulla biodisponibilità. La pomalidomide può essere somministrata indipendentemente dall'assunzione di cibo.
Distribuzione
Il volume di distribuzione allo steady state è compreso tra 62 e 138 l. La concentrazione nel liquido seminale di soggetti sani è pari a circa il 67% del livello plasmatico. Il legame proteico è basso.
Metabolismo
La pomalidomide è il principale componente in circolo (circa il 70% della radioattività plasmatica). Non sono stati rilevati metaboliti che corrispondessero a >10% della radioattività totale nel plasma.
Le principali vie metaboliche della radioattività escreta sono idrossilazione con successiva glucuronidazione e idrolisi. In vitro, CYP1A2 e CYP3A4 sono stati identificati come i principali enzimi coinvolti nell'idrossilazione della pomalidomide mediata dal CYP, con ulteriori contributi di minore entità del CYP2C19 e CYP2D6.
In 14 volontari maschi sani che dopo una dose orale singola di 4 mg di pomalidomide hanno fumato 25 sigarette al giorno per 10 giorni si è osservato un aumento del 14% della Cmax e una riduzione del 32% dell'AUC della pomalidomide, rispetto a 13 volontari maschi sani non fumatori.
Eliminazione
L'emivita di eliminazione è di circa 9,5 ore nei soggetti sani e di circa 7,5 ore nei pazienti con mieloma multiplo. La clearance corporea totale (CL/F) media è di circa 7‑10 l/h.
Il 73% della dose viene eliminato nelle urine e il 15% nelle feci, con circa il 2% e l'8% di pomalidomide escreta rispettivamente nelle urine e nelle feci.
Cinetica di gruppi di pazienti speciali
Disturbi della funzionalità epatica
I parametri farmacocinetici sono risultati lievemente alterati nei pazienti con compromissione della funzionalità epatica (in base ai criteri di Child-Pugh), rispetto ai soggetti sani. L'esposizione media alla pomalidomide è aumentata del 51% (IC al 90%: 9%, 110%) nei pazienti con compromissione della funzionalità epatica lieve, rispetto ai soggetti sani. L'esposizione media alla pomalidomide è aumentata del 58% (IC al 90%: 13%, 119%) nei pazienti con compromissione della funzionalità epatica moderata, rispetto ai soggetti sani. L'esposizione media alla pomalidomide è aumentata del 72% (IC al 90%: 24%, 138%) nei pazienti con compromissione della funzionalità epatica grave, rispetto ai soggetti sani.
Disturbi della funzionalità renale
Le analisi di farmacocinetica di popolazione hanno mostrato che i parametri farmacocinetici della pomalidomide non sono stati influenzati in misura apprezzabile nei pazienti con disturbi della funzionalità renale (definita in base alla clearance della creatinina o al tasso di filtrazione glomerulare stimato [eGFR]), rispetto ai pazienti con funzionalità renale nella norma (CrCl ≥60 ml/min). L'esposizione media normalizzata AUC alla pomalidomide è stata del 98,2% (IC al 90%: 77,4%, 120,6%) nei pazienti con insufficienza renale moderata (eGFR ≥30 e ≤45 ml/min/1,73 m2), rispetto ai pazienti con funzionalità renale nella norma. L'esposizione media normalizzata AUC alla pomalidomide è stata del 100,2% (IC al 90%: 79,7%, 127,0%) nei pazienti con insufficienza renale grave che non richiedevano dialisi (CrCl <30 ml/min o eGFR <30 ml/min/1,73 m2), rispetto ai pazienti con funzionalità renale nella norma. L'esposizione media normalizzata AUC alla pomalidomide è aumentata del 35,8% (IC al 90%: 7,5%, 70,0%) nei pazienti con insufficienza renale grave che richiedevano dialisi (CrCl <30 ml/min con necessità di dialisi), rispetto ai pazienti con funzionalità renale nella norma.
Pazienti anziani
Nei pazienti di età compresa tra 61 e 82 anni, i valori medi dei parametri farmacocinetici AUC (0-∞) e Cmax sono risultati in generale simili a quelli dei pazienti più giovani.
Bambini e adolescenti
Non sono disponibili dati sulla farmacocinetica nei pazienti pediatrici.
Dati preclinici
Genotossicità/cancerogenicità
La pomalidomide non è risultata mutagena nei saggi di mutazione in batteri e cellule di mammifero e non ha indotto aberrazioni cromosomiche nei linfociti del sangue periferico umano, né formazione di micronuclei negli eritrociti policromatici nel midollo osseo di ratti trattati con dosi fino a 2000 mg/kg/die. Non sono stati effettuati studi di cancerogenicità.
Fertilità e primo sviluppo embrionale
In uno studio sulla fertilità e sul primo sviluppo embrionale nei ratti, la pomalidomide è stata somministrata a maschi e femmine a dosi di 25, 250 e 1000 mg/kg/die. L'esame dell'utero al 13° giorno di gestazione ha evidenziato una riduzione del numero medio di embrioni vitali e un aumento della perdita post-impianto a tutti i livelli di dosaggio. Pertanto, il NOAEL per questi effetti osservati è stato <25 mg/kg/die (AUC24h – 39'960 ng•h/ml; esposizione 99 volte più elevata alla più bassa dose testata, rispetto a una dose di 4 mg). Quando i maschi trattati in questo studio si sono accoppiati con femmine non trattate, tutti i parametri uterini erano paragonabili ai controlli. Sulla base di questi risultati, gli effetti osservati sono stati attribuiti al trattamento delle femmine.
Sviluppo embrio-fetale
La pomalidomide si è rivelata teratogena sia nei ratti che nei conigli, quando è stata somministrata durante il periodo dell'organogenesi principale. Nello studio di tossicità dello sviluppo embriofetale nel ratto, sono stati osservati, a tutti i livelli di dosaggio (25, 250 e 1000 mg/kg/die), malformazioni o assenza della vescica urinaria, assenza della tiroide e fusione ed errato allineamento degli elementi vertebrali lombari e toracici (arco vertebrale e/o neurale). In questo studio non è stata osservata tossicità materna. Pertanto, il NOAEL materno era pari a 1000 mg/kg/die e il NOAEL per la tossicità dello sviluppo era pari a <25 mg/kg/die (AUC24h = 34'340 ng•h/ml il 17° giorno di gestazione; esposizione 85 volte più elevata alla più bassa dose testata, rispetto a una dose di 4 mg). Nel coniglio, la pomalidomide a dosi comprese tra 10 e 250 mg/kg ha prodotto malformazioni dello sviluppo embrio-fetale. Un aumento delle anomalie cardiache è stato osservato a tutte i livelli di dose, con aumenti significativi a 250 mg/kg/die. A 100 e 250 mg/kg/die vi sono stati leggeri aumenti delle perdite post-impianto e leggere riduzioni del peso corporeo fetale. A 250 mg/kg/die, le malformazioni fetali hanno riguardato anomalie degli arti (arti anteriori e/o posteriori flessi e/o ruotati, dita non fissate o assenti) e malformazioni scheletriche associate (mancata ossificazione del metacarpo, disallineamento di falange e metacarpo, dita assenti, mancata ossificazione della falange e tibia breve non ossificata o ricurva); moderata dilatazione del ventricolo laterale nel cervello; posizionamento anomalo dell'arteria succlavia destra; lobo intermedio assente nei polmoni; rene abbassato; alterata morfologia del fegato; incompleta o mancata ossificazione della pelvi; aumento della media di coste toraciche soprannumerarie e riduzione della media di ossa tarsali ossificate. Leggera riduzione dell'aumento ponderale materno, significativa riduzione dei trigliceridi e significativa riduzione del peso assoluto e relativo della milza sono state osservate a 100 e 250 mg/kg/die. Il NOAEL materno era pari a 10 mg/kg/die e il NOAEL dello sviluppo era <10 mg/kg/die (AUC24h = 418 ng·h/ml il 19° giorno di gestazione; 1 volta l'esposizione clinica alla dose di 4 mg).
Altre indicazioni
Stabilità
Il medicamento non deve essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Indicazioni particolari concernenti l'immagazzinamento
Conservare nella confezione originale, a temperature non superiori a 25 °C e fuori dalla portata dei bambini.
Indicazioni per la manipolazione
Come per i citostatici, anche la manipolazione e lo smaltimento di Imnovid richiedono particolare cautela (vedere anche «Posologia / impiego»).
Le donne potenzialmente fertili che manipolano la pomalidomide devono indossare guanti protettivi.
Le capsule non devono essere aperte né frantumate. Se la polvere di pomalidomide viene a contatto con la cute, lavare la cute immediatamente e accuratamente con acqua e sapone. Se la pomalidomide viene a contatto con le mucose, sciacquare accuratamente con acqua.
Numero dell'omologazione
61249 (Swissmedic)
Titolare dell’omologazione
Celgene GmbH, Zurigo
Stato dell'informazione
Gennaio 2020
Composition
Principes actifs
Pomalidomidum.
Excipients
Mannitol, amidon prégélatinisé, fumarate de stéaryle sodique.
Enveloppe des gélules: gélatine, dioxyde de titane, oxyde de fer jaune (uniquement pour les gélules dures 1 mg, 2 mg et 3 mg), indigocarmine E132, érythrosine E127 (uniquement pour les gélules dures 2 mg), bleu brillant FCF E133 (uniquement pour les gélules dures 4 mg).
Encre d'impression: gomme laque, oxyde de fer noir, dioxyde de titane, siméticone, propylène glycol, solution d'ammoniaque.
Une gélule dure contient au maximum 0,018 mg (gélules dures 1 mg) ou au maximum 0,036 mg (gélules dures 2 mg) ou au maximum 0,025 mg (gélules dures 3 mg) ou au maximum 0,033 (gélules dures 4 mg) de sodium.
Forme pharmaceutique et quantité de principe actif par unité
Gélules de 1 mg, 2 mg, 3 mg et 4 mg.
Indications/Possibilités d’emploi
Imnovid, en association avec le bortézomib et la dexaméthasone, est indiqué pour le traitement du myélome multiple (MM) chez les patients adultes ayant déjà reçu au moins un traitement antérieur comportant le lénalidomide.
Imnovid, en association avec la dexaméthasone, est indiqué pour le traitement du myélome multiple en rechute et réfractaire chez les patients ayant déjà reçu au moins deux traitements antérieurs (comportant le lénalidomide et le bortézomib) et dont la maladie a progressé pendant le dernier traitement.
Posologie/Mode d’emploi
Le traitement doit être initié et surveillé par un hématologue ou un oncologue expérimenté.
Imnovid en association avec le bortézomib et la dexaméthasone (PVd) chez les patients atteints d'un myélome multiple, ayant déjà reçu au moins un traitement antérieur.
La dose initiale recommandée d'Imnovid est de 4 mg par voie orale une fois par jour aux jours 1 à 14 d'un cycle répété de traitement de 21 jours.
La dose recommandée de bortézomib est de 1,3 mg/m2 et la dose recommandée de dexaméthasone est de 20 mg/jour par voie orale une fois par jour, l'administration devant suivre le schéma posologique indiqué au tableau 1.
La posologie est maintenue ou modifiée en fonction des résultats des examens cliniques et des analyses biologiques. Le traitement doit être interrompu en cas de progression de la maladie.
Tableau 1: Schéma posologique recommandé pour Imnovid en association avec le bortézomib et la dexaméthasone
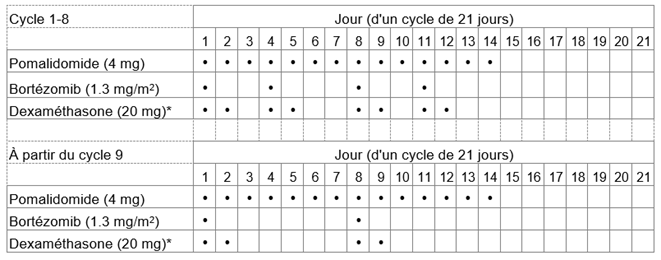
* Pour les patients âgés de >75 ans, voir rubrique «Instructions spéciales pour la posologie».
Imnovid en association avec la dexaméthasone (Pd) chez les patients présentant un myélome multiple en rechute et réfractaire, ayant déjà reçu au moins deux traitements antérieurs
La dose initiale recommandée d'Imnovid est de 4 mg par voie orale une fois par jour aux jours 1 à 21 d'un cycle répété de traitement de 28 jours jusqu'à la progression de la maladie. La dose recommandée de dexaméthasone est de 40 mg une fois par jour aux jours 1, 8, 15 et 22 de chaque cycle de traitement de 28 jours.
La posologie est maintenue ou modifiée en fonction des résultats des examens cliniques et des analyses biologiques.
Le schéma posologique est indiqué au tableau 2.
Tableau 2: Schéma posologique recommandé pour Imnovid en association avec la dexaméthasone
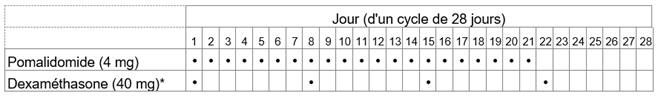
* Pour les patients âgés de >75 ans, voir rubrique «Instructions spéciales pour la posologie».
Ajustement de la posologie
Hématotoxicité
En cas de thrombocytopénie avec chute de la numération plaquettaire <25 x 109/l ou en cas de neutropénie avec chute des polynucléaires neutrophiles (PNN) <0,5 x 109/l ou en cas de neutropénie fébrile (fièvre ≥38,5 °C et PNN <1,0 x 109/l), le traitement par pomalidomide doit être interrompu et suivi d'un contrôle hebdomadaire de la numération formule sanguine complète (et en cas de chute des polynucléaires neutrophiles, l'utilisation de facteurs de croissance (G-CSF) doit être envisagée à l'appréciation du médecin traitant). Après le retour à la normale de la numération des thrombocytes/du taux des polynucléaires neutrophiles, le traitement par le pomalidomide doit être repris à la dose de 3 mg par jour. Pour chaque chute ultérieure (<25 x 109/l et <0,5 × 109/l respectivement), le traitement par le pomalidomide doit être interrompu. Après le retour à la normale de la numération des thrombocytes/du taux des polynucléaires neutrophiles, le traitement par le pomalidomide doit être repris à une dose inférieure de 1 mg à la dose antérieure.
Imnovid en association avec le bortézomib et la dexaméthasone (PVd): pour commencer un nouveau cycle de pomalidomide, le taux des polynucléaires neutrophiles doit être ≥ 1 × 109/l et la numération des plaquettes ≥50 × 109/l.
Imnovid en association avec la dexaméthasone (Pd): pour commencer un nouveau cycle de pomalidomide, le taux des polynucléaires neutrophiles doit être ≥0.5 x 109/l et la numération des plaquettes ≥50 x 109/l.
Autres effets indésirables de grade 3/4
En cas d'autres effets indésirables de grade 3/4, jugés comme étant liés au pomalidomide, le traitement doit être interrompu et repris à une dose inférieure de 1 mg à la dose antérieure après résolution de l'effet indésirable à un grade ≤2, à l'appréciation du médecin. Si les effets indésirables réapparaissent après réduction de la dose à 1 mg, le médicament doit être arrêté.
Pour des précisions relatives aux ajustements posologiques en raison d'une toxicité au bortézomib, il convient de se référer à l'information destinée aux professionnels du médicament concerné.
Ajustement posologique en cas d'administration concomitante d'inhibiteurs du CYP1A2
Il convient d'éviter l'utilisation concomitante du pomalidomide avec des inhibiteurs puissants du CYP1A2. Envisager d'autres modes de traitement. En cas d'administration concomitante d'inhibiteurs puissants du CYP1A2 (p.ex. ciprofloxacine et fluvoxamine) avec le pomalidomide, la dose de pomalidomide doit être réduite de 50%.
Arrêt de pomalidomide
L'interruption ou l'arrêt du traitement par le pomalidomide doit être envisagé(e) en cas d'éruption cutanée de grade 2 ou 3.
Pomalidomide doit être arrêté en cas d'angioedème, d'anaphylaxie, d'éruption cutanée de grade 4, de dermatite exfoliative ou d'éruption bulleuse, ou de suspicion de syndrome de Stevens-Johnson (SJS), de syndrome de Lyell (TEN) ou d'une réaction médicamenteuse accompagnée d'une éosinophilie et de symptômes systémiques (DRESS). Après l'interruption en raison de ces réactions, le traitement ne doit pas être repris.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Chez les patients présentant une insuffisance hépatique légère à modérée (grade A ou B de la classification de Child-Pugh), la dose initiale recommandée est de 3 mg par jour (réduction de la dose de 25%). Chez les patients présentant une insuffisance hépatique sévère (grade C de la classification de Child-Pugh), la dose recommandée est de 2 mg (réduction de la dose de 50%).
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique du pomalidomide n'est nécessaire chez les patients présentant une insuffisance rénale modérée et chez ceux présentant une insuffisance rénale sévère ne nécessitant pas de dialyse. Chez les patients présentant une insuffisance rénale sévère et nécessitant des dialyses, la dose initiale recommandée est de 3 mg par jour (réduction de la dose de 25%). Les jours d'hémodialyse, le pomalidomide doit être pris après l'hémodialyse.
Patients âgés
Aucune adaptation de la dose de pomalidomide n'est nécessaire.
Imnovid en association avec le bortézomib et la dexaméthasone (PVd) après au moins un traitement antérieur
Chez les patients âgés de >75 ans, la dose de dexaméthasone est de 10 mg une fois par jour aux jours 1, 2, 4, 5, 8, 9, 11 et 12 d'un cycle de 21 jours pour les cycles 1-8, et à partir du cycle 9, de 10 mg une fois par jour aux jours 1, 2, 8 et 9 d'un cycle de 21 jours.
Imnovid en association avec la dexaméthasone (Pd) après au moins 2 traitements antérieurs
Chez les patients âgés de plus de 75 ans, la dose initiale de dexaméthasone est de 20 mg un fois par jour aux jours 1, 8, 15 et 22 de chaque cycle de 28 jours.
Enfants et adolescents
Le pomalidomide n'a pas été étudié en pédiatrie.
Mode d'administration
Imnovid doit être pris chaque jour par voie orale environ à la même heure. Les gélules ne doivent pas être ouvertes, cassées ou mâchées. Les gélules d'Imnovid doivent être avalées entières, de préférence avec de l'eau, indépendamment des repas. Si le patient oublie de prendre une dose d'Imnovid pendant une journée, il doit prendre la dose normale prescrite à l'heure habituelle le lendemain. La dose ne doit pas être ajustée pour compenser une dose d'Imnovid omise le jour précédent.
Pour des précisions relatives à d'autres médicaments utilisés en association avec le pomalidomide, il convient de se référer à l'information destinée aux professionnels du médicament concerné.
Contre-indications
Hypersensibilité au pomalidomide, à l'un des excipients, ou encore au thalidomide et au lénalidomide.
Grossesse.
Femmes en âge de procréer, sauf si toutes les conditions du programme de prévention de la grossesse sont remplies.
Pour des précisions relatives à d'autres médicaments utilisés en association avec le pomalidomide, il convient de se référer à l'information destinée aux professionnels du médicament concerné.
Mises en garde et précautions
Le pomalidomide est un analogue du thalidomide. Le thalidomide a des effets tératogènes connus chez l'homme, lesquels provoquent des anomalies congénitales graves menaçant le pronostic vital de l'enfant à naître. Chez le rat et le lapin, le pomalidomide s'est avéré tératogène lorsqu'il a été administré pendant la phase d'organogenèse majeure (voir «Données précliniques»). Si le pomalidomide est pris pendant la grossesse, il faut s'attendre à un effet tératogène chez l'homme.
Programme de prévention de la grossesse
Programme pour les patientes
Les conditions du programme de prévention de la grossesse doivent être remplies par toutes les patientes, à moins de pouvoir certifier que la patiente est dans l'impossibilité de procréer.
Critères pour l'évaluation du potentiel de procréer
Toute patiente ou conjointe de patient est considérée comme capable de procréer, à moins qu'elle ne remplisse au moins l'une des conditions suivantes:
- Âge ≥50 ans et aménorrhée naturelle depuis ≥1 an*
- Déficience ovarienne prématurée avérée
- Antécédent de salpingo-oophorectomie bilatérale, de ligature des trompes bilatérale ou d'hystérectomie
- Génotype XY, syndrome de Turner, aplasie de l'utérus
* Une aménorrhée après un traitement anticancéreux n'exclut pas la capacité de procréer.
Consultation
Le pomalidomide est contre-indiqué chez les femmes capables de procréer qui ne remplissent pas toutes les conditions ci-dessous:
- La patiente comprend le risque tératogène attendu auquel serait exposé un enfant à naître.
- Elle comprend la nécessité d'une contraception fiable sans interruption commencée 4 semaines avant le début du traitement, poursuivie pendant toute la durée du traitement, pauses de traitement comprises, et pendant les 4 semaines suivant la fin du traitement.
- Même en cas d'aménorrhée, toute patiente en âge de procréer doit respecter strictement toutes les recommandations fournies pour une contraception efficace.
- Elle doit être capable de respecter les exigences d'une méthode de contraception fiable.
- Elle est informée et a compris les conséquences d'une grossesse ainsi que la nécessité de consulter rapidement le médecin si une grossesse est suspectée.
- Elle comprend la nécessité de faire des tests de grossesse toutes les 4 semaines et accepte de s'y soumettre.
- Elle a confirmé avoir compris les risques et mesures de sécurité nécessaires dans le cadre d'un traitement par le pomalidomide.
Chez les femmes en mesure de procréer, le médecin prescripteur doit s'assurer que
- La patiente remplit les conditions indiquées ci-dessus.
- La patiente remplit les conditions spécifiées par le programme de prévention de la grossesse, y compris confirmation d'une compréhension suffisante.
- La patiente a appliqué des mesures contraceptives suffisantes au moins 4 semaines avant le début du traitement, continue à appliquer des mesures contraceptives suffisantes pendant toute la durée du traitement, pauses de traitement comprises, et continuera à les appliquer encore au moins 4 semaines au-delà de la fin du traitement. Chez les patientes nécessitant un traitement immédiat par le pomalidomide, une contraception adéquate, associée à l'utilisation de préservatifs, doit être assurée pendant les 7 jours précédant le début du traitement.
- Un test de grossesse fait avant le traitement soit négatif.
Contraception
Les femmes en mesure de procréer doivent appliquer une méthode de contraception fiable pendant les 4 semaines précédant le début du traitement, pendant toute la durée du traitement, pauses de traitement comprises, et pendant les 4 semaines suivant la fin du traitement. Chez les patientes nécessitant un traitement immédiat par le pomalidomide, une contraception adéquate, associée à l'utilisation de préservatifs, doit être assurée pendant les 7 jours précédant le début du traitement. Si une méthode de contraception fiable n'a pas été utilisée auparavant, la patiente doit être adressée à un service médical de consultation capable de conseiller et informer la patiente de façon approfondie pour le choix d'une méthode contraceptive fiable.
Les méthodes de contraception suivantes peuvent être considérées comme fiables:
- Méthodes indépendantes de la patiente:
- Implant
- Acétate de médroxyprogestérone retard
- Stérilisation
- Méthodes dépendantes de la patiente:
- Renoncement total aux rapports hétérosexuels
- Rapports hétérosexuels uniquement avec un partenaire stérilisé par une vasectomie attestée par deux examens confirmant l'absence de spermatozoïdes.
- Contraceptifs oraux à la progestérone seule.
En raison du risque accru de thromboembolies veineuses sous pomalidomide, les contraceptifs oraux œstro-progestatifs ne sont pas recommandés. Si une patiente utilise déjà des contraceptifs oraux œstro-progestatifs, il convient de considérer le passage à une autre méthode de contraception. Le risque de thromboembolies veineuses persiste pendant 4 à 6 semaines après l'arrêt du contraceptif oral œstro-progestatif. Si aucune autre méthode ne peut être utilisée, on considérera une prévention antithrombotique pendant la durée d'utilisation du contraceptif oral œstro-progestatif. La patiente doit être dûment informée du risque de thromboembolie veineuse.
Les dispositifs intra-utérins (stérilets) sont associés à un risque accru d'infections lors de la mise en place et peuvent provoquer des saignements vaginaux ou règles irrégulières. Ces méthodes ne sont donc pas recommandées.
Test de grossesse
Des tests de grossesse avec une sensibilité d'au moins 25 UI/ml hCG doivent être effectués chez les femmes capables de procréer.
Chaque cas d'une patiente dont le test de grossesse est positif doit, sans attendre, être reporté au Swiss Teratogen Information Service (STIS) à Lausanne, au moyen du formulaire Swissmedic «Annonce d'effets indésirables suspectés d'un médicament (EI)».
- Avant le début du traitement
Un test de grossesse sous contrôle médical doit être réalisé lors de la consultation où le pomalidomide est prescrit ou au cours des trois jours suivant la visite chez le médecin prescripteur. À la date du test, la patiente doit avoir appliqué une méthode contraceptive fiable depuis au moins 4 semaines. Le test doit assurer que la patiente n'est pas enceinte au début du traitement par le pomalidomide.
- Avant le début du traitement chez les patientes nécessitant un traitement immédiat
Une détermination quantitative du taux sérique de hCG doit être faite immédiatement. Après 7 jours d'application d'une méthode de contraception efficace, associée à l'utilisation de préservatifs, le test doit être répété. Si les deux tests confirment que la patiente n'est pas enceinte, le traitement peut être commencé.
- Pendant et après le traitement
Un test de grossesse doit être fait toutes les 4 semaines pendant le traitement, et 4 semaines après la fin du traitement. Ces tests de grossesse doivent être faits pendant les visites chez le médecin pour la prescription du pomalidomide ou dans les trois jours précédant une telle visite.
Les tests de grossesse et la prescription et remise du pomalidomide doivent de préférence avoir lieu le même jour. Le pomalidomide doit être remise au plus tard sous 7 jours à compter de la date de prescription.
Programme chez les patients de sexe masculin
Chez les patients de sexe masculin, les données cliniques attestent du passage de la substance dans le sperme pendant la prise d'Imnovid. Les patients dont la partenaire est en mesure de procréer doivent de ce fait utiliser des préservatifs lors des rapports sexuels pendant le traitement par Imnovid et au moins pendant les 7 jours après l'arrêt du traitement. Les hommes traités par Imnovid doivent remplir les conditions suivantes:
- Ils doivent avoir compris le risque tératogène attendu s'ils ont des rapports sexuels avec une femme en mesure de procréer.
- Ils doivent avoir compris et accepté qu'ils doivent utiliser des préservatifs pour tous les rapports sexuels avec une femme en mesure de procréer pendant toute la durée du traitement, pauses de traitement comprises, et pendant les 7 jours suivant la fin du traitement.
Le médecin prescripteur doit assurer que les patients de sexe masculin ont compris et accepté d'utiliser des préservatifs pendant toute la durée du traitement, pauses du traitement compris, et pendant les 7 jours suivant la fin du traitement, s'ils ont des rapports sexuels avec une femme en mesure de procréer.
Les patients ne doivent en aucun cas faire un don de sperme pendant leur traitement par Imnovid ou pendant les 7 jours suivant la fin de ce traitement.
Précautions supplémentaires
Les patients et patientes doivent être instruits de ne jamais donner ce médicament à d'autres personnes et de rapporter les gélules non utilisées à leur médecin ou à leur pharmacien à la fin du traitement.
Documents d'information
- Brochure d'information pour les patients et patientes relative au programme de prévention de la grossesse du pomalidomide avec formulaire de consentement des patients et patientes
- Mesures contraceptives à prendre
- Brochure d'information destinée aux professionnels de la santé relative au programme de prévention de la grossesse du pomalidomide
Autres mises en garde et précautions
Effets indésirables hématologiques
Les patients doivent être surveillés pour détecter l'apparition d'effets indésirables hématologiques, en particulier d'une neutropénie. L'hémogramme complet doit être surveillé une fois par semaine pendant les 8 premières semaines, puis une fois par mois. Une modification de dose peut être nécessaire (voir rubrique «Posologie/Mode d'emploi»).
Evénements thrombo-emboliques
On a rapporté des événements thrombo-emboliques veineux (principalement des thromboses veineuses profondes et des embolies pulmonaires). C'est pourquoi, un traitement anticoagulant est conseillé (sauf s'il est contre-indiqué). La décision de mettre en place des mesures prophylactiques anti-thrombotiques devra être prise après une évaluation attentive des facteurs de risque sous-jacents propres à chaque patient.
Affections cardiaques
Des cas d'insuffisance cardiaque, y compris d'insuffisance cardiaque congestive, d'œdème pulmonaire et de fibrillation auriculaire ont été rapportés, surtout chez les patients ayant présenté une affection cardiaque antérieure ou des facteurs de risque cardiaque. Lorsqu'il est envisagé de traiter ces patients par le pomalidomide, la prudence s'impose ainsi qu'une surveillance régulière des signes et symptômes d'une insuffisance cardiaque.
Syndrome de lyse tumorale
Un syndrome de lyse tumorale peut survenir. Les patients ayant le plus grand risque de syndrome de lyse tumorale sont ceux qui ont une charge tumorale élevée avant le traitement. Ces patients doivent faire l'objet d'une surveillance étroite, et des précautions appropriées doivent être prises.
Réactions allergiques et réactions cutanées sévères
Des cas d'angiœdème, d'anaphylaxie et de réactions cutanées sévères, notamment le syndrome de Stevens-Johnson (SJS), le syndrome de Lyell (nécrolyse épidermique toxique - TEN), et une réaction médicamenteuse accompagnée d'une éosinophilie et de symptômes systémiques (DRESS), ont été rapportés. Le DRESS peut se manifester sous forme d'éruption cutanée ou de dermatite exfoliative associée à une éosinophilie, une fièvre et/ou une lymphadénopathie avec complications systémiques comme une hépatite, une néphrite, une pneumopathie inflammatoire, une myocardite et/ou une péricardite. Ces événements peuvent être fatals. L'interruption ou l'arrêt du traitement par le pomalidomide doit être envisagé(e) en cas d'éruption cutanée de grade 2 ou 3. Pomalidomide doit être arrêté en cas d'angiœdème, d'anaphylaxie, d'éruption cutanée de grade 4, de dermatite exfoliative ou d'éruption bulleuse, ou de suspicion d'un SJS, d'une TEN ou d'un DRESS. Après l'interruption en raison de ces réactions, le traitement ne doit pas être repris.
Étourdissements et confusion
Des étourdissements et une confusion ont été rapportés. Il convient d'indiquer aux patients les situations dans lesquelles les étourdissements ou la confusion peuvent constituer un problème.
Cancers secondaires
Des cancers secondaires ont été rapportés chez des patients recevant le pomalidomide. Le médecin doit examiner soigneusement les patients avant et pendant le traitement en utilisant les méthodes habituelles de dépistage des cancers pour surveiller le développement de cancers secondaires et instaurer un traitement le cas échéant.
Troubles de la fonction hépatique
Des élévations importantes des taux d'alanine-aminotransférase et de bilirubine sériques ont été observées chez les patients traités par le pomalidomide (voir rubrique «Effets indésirables»). Il existe également des cas d'hépatite, y compris de réactivation de l'hépatite B, qui ont donné lieu à l'arrêt du pomalidomide. Un contrôle régulier de la fonction hépatique est conseillé.
Infections
Des infections (24,0 à 30,9% de grade 3 ou 4) se sont présentées chez 55,0 à 80,2% des patients qui avaient reçu un traitement d'association avec le pomalidomide dans des études cliniques. Des infections des voies respiratoires supérieures et des pneumonies étaient les infections qui sont survenues le plus fréquemment. Des infections à issue fatale (grade 5) sont survenues chez 2,7 à 4,0% des patients. Chez 2,0 à 2,9% des patients, les infections ont conduit à l'arrêt du pomalidomide.
Les patients présentant des facteurs de risque connus de survenue d'infections doivent être étroitement surveillés. Il convient de conseiller à tous les patients de contacter immédiatement un médecin dès le premier signe d'une infection (p.ex. toux, fièvre, etc.), afin d'en diminuer si possible la sévérité par un traitement précoce.
De rares cas de réactivation de l'hépatite B ont été rapportés à la suite du traitement par le pomalidomide en association avec la dexaméthasone chez des patients présentant des antécédents d'infection par le virus de l'hépatite B (VHB). Certains de ces cas ont évolué vers une insuffisance hépatique aiguë et ont conduit à l'arrêt du traitement par le pomalidomide. La sérologie VHB doit être déterminée avant l'initiation du traitement par le pomalidomide. Chez les patients ayant un résultat positif au dépistage du virus de l'hépatite B, une consultation chez un médecin spécialisé dans le traitement de l'hépatite B est recommandée. La prudence s'impose en cas d'administration de pomalidomide en association avec la dexaméthasone chez des patients préalablement infectés par le VHB. Ces patients doivent être étroitement surveillés tout au long du traitement afin de détecter les signes et symptômes d'infection active par le VHB.
Troubles rénaux
Dans des études cliniques, on a observé un taux plus élevé d'effets indésirables hématologiques (anémie et thrombocytopénie) et rénaux (lésions rénales aiguës) (voir aussi «Propriétés/Effets») chez les patients dont la clairance de la créatinine était ≤45 ml/min, et ayant reçu le pomalidomide en association avec le bortézomib et la dexaméthasone. Ces patients doivent être soigneusement surveillés.
Pour des précisions relatives à d'autres médicaments utilisés en association avec le pomalidomide, il convient de se référer à l'information destinée aux professionnels du médicament concerné.
Ce médicament renferme moins de 1 mmol de sodium (23 mg) par gélule, ce qui signifie qu'il s'agit d'un médicament pratiquement «sans sodium».
Interactions
Interactions éventuelles d'autres médicaments avec le pomalidomide
Le pomalidomide est métabolisé en partie par les CYP1A2 et CYP3A4/5. C'est également un substrat de la glycoprotéine P. L'administration concomitante de pomalidomide avec des médicaments comme par exemple, le kétoconazole, un inhibiteur puissant du CYP3A4/5 et de la P-gp, ou avec la carbamazépine, un inducteur puissant du CYP3A4/5, n'a pas eu d'effets cliniquement pertinents sur l'exposition au pomalidomide.
L'administration concomitante de fluvoxamine, un inhibiteur puissant du CYP1A2, et de pomalidomide en présence de kétoconazole a augmenté de 107% l'exposition moyenne au pomalidomide intervalle de confiance à 90% [91% à 124%] par rapport au pomalidomide plus kétoconazole seuls. L'utilisation concomitante de fluvoxamine (un inhibiteur puissant du CYP1A2) et du pomalidomide a augmenté de 125% l'exposition moyenne au pomalidomide intervalle de confiance à 90% [98% à 157%] par rapport au pomalidomide seul chez les volontaires sains. La dose de pomalidomide doit être réduite de 50% en cas d'administration concomitante d'inhibiteurs puissants du CYP1A2 (p.ex. ciprofloxacine et fluvoxamine) avec le pomalidomide.
Interactions éventuelles du pomalidomide avec d'autres médicaments
Le pomalidomide n'inhibe par les CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 ou CYP3A4/5 in vitro. En outre, in vitro, le pomalidomide n'est pas un inducteur des enzymes CYP1A2, CYP2B6, CYP2C9, CYP2C19 et CYP3A4/5.
Le pomalidomide n'est pas un inhibiteur de la glycoprotéine P (P-gp) et n'a montré dans les études in vitro aucun ou un faible effet inhibiteur sur la «Breast Cancer Resistant Protein» (BCRP), les transporteurs «Organic Anion Transporter Protein» (OATP) 1B1, OATP1B3, «Organic Cation Transporter» OAT1 et OAT3 ainsi que sur le transporteur «Organic Anion Transporter Protein» OCT2.
On ne s'attend pas à ce que le pomalidomide provoque des interactions pharmacocinétiques cliniquement pertinentes en raison d'une inhibition ou induction ou d'une inhibition des transporteurs lorsqu'il est administré en association avec des substrats de ces enzymes ou des transporteurs. Le potentiel de telles interactions médicamenteuses ainsi que l'effet possible du pomalidomide sur la pharmacocinétique des contraceptifs oraux, n'ont pas été évalués dans le cadre d'études cliniques.
Grossesse/Allaitement
Grossesse
Un effet tératogène du pomalidomide est attendu chez l'être humain. Le pomalidomide est contre-indiqué pendant la grossesse et ne doit pas être utilisé chez des femmes en âge de procréer à moins que toutes les conditions relatives à la contraception soient remplies (voir la rubrique «Mises en garde et précautions»).
Allaitement
On ignore si le pomalidomide est excrété dans le lait maternel. Après administration chez des rates allaitantes, le pomalidomide a été détecté dans le lait. Compte tenu du risque d'effets indésirables du pomalidomide chez l'enfant allaité, une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre le traitement en prenant en compte l'importance du traitement par le pomalidomide pour la mère.
Effet sur l’aptitude à la conduite et l’utilisation de machines
Aucune étude relative à l'effet du pomalidomide sur l'aptitude à la conduite et l'utilisation de machines n'a été réalisée. Des cas de syncope, fatigue et de vertiges ont été signalés lors de l'utilisation du pomalidomide. C'est pourquoi, la prudence est conseillée aux patients sous pomalidomide s'ils conduisent des véhicules ou utilisent des machines.
Effets indésirables
Imnovid en association avec le bortézomib et la dexaméthasone (PVd) chez des patients présentant un myélome multiple, ayant reçu au moins un traitement antérieur
Dans l'étude randomisée CC-4047-MM-007, 278 patients ont reçu le pomalidomide, le bortézomib et la dexaméthasone.
Les maladies du sang et du système lymphatique les plus fréquemment rapportées étaient la neutropénie, la thrombocytopénie et l'anémie. L'effet indésirable le plus fréquemment rapporté était la neuropathie périphérique sensitive. L'effet indésirable sévère le plus fréquemment rapporté était la pneumonie (11,5%).
Imnovid en association avec la dexaméthasone (Pd) chez les patients présentant un myélome multiple en rechute et réfractaire, ayant déjà reçu au moins deux traitements antérieurs
Dans trois études cliniques (CC-4047-MM-003, CC-4047-MM-002 et CC-4047-IFM-2009-02), 455 patients ont été exposés à 4 mg de pomalidomide.
Les effets indésirables les plus fréquemment rapportés étaient neutropénie, anémie, thrombocytopénie, fatigue, fièvre, constipation et diarrhée.
L'effet indésirable grave le plus fréquemment rapporté était la pneumonie (12,5%).
Les fréquences sont définies comme suit: très fréquent (≥1/10); fréquent (<1/10, ≥1/100); occasionnel (<1/100, ≥1/1000); rares (1/1000, ≥1/10'000); très rares (<1/10'000).
Infections et infestations#
Très fréquent: infections des voies respiratoires supérieures (20,9%), pneumonie (19,1%), bronchite (14,0%), infection virale des voies respiratoires supérieures (11,2%).
Fréquent: septicémie, choc septique, colite à Clostridium difficile, bronchopneumonie, grippe, infection des voies urinaires, infection des voies respiratoires, infections des voies respiratoires basses, bronchiolite, infection pulmonaire, sinusite, rhinopharyngite, candidose, candidose orale.
Occasionnel: septicémie neutropénique, herpès zoster*.
Rare: réactivation du virus de l'hépatite B*.
# = Tous les termes préférentiels de la classe de système/organe des infections (y compris les infections bactériennes, virales et fongiques) sont mentionnés, à l'exception des infections rares d'intérêt sanitaire public.
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes)
Fréquent: carcinome baso-cellulaire.
Occasionnel: carcinome épidermoïde de la peau*.
Affections hématologiques et du système lymphatique
Très fréquent: neutropénie (47,9%), anémie (44,0%), thrombocytopénie (36,7%), leucopénie (13,0%).
Fréquent: neutropénie fébrile, lymphopénie, diminution du nombre de globules blancs, diminution du taux des polynucléaires neutrophiles, diminution de la numération des thrombocytes.
Occasionnel: pancytopénie*.
Affections du système immunitaire
Occasionnel: réactions allergiques (p.ex. angioedème, urticaire)*.
Rare: anaphylaxie*.
Affections endocriniennes
Rare: hypothyroïdie*.
Troubles du métabolisme et de la nutrition
Très fréquent: hypokaliémie (15,5%), hyperglycémie (14,4%), perte de l'appétit (11,6%).
Fréquent: déshydratation, hyponatrémie, hyperkaliémie, hypercalcémie, hypocalcémie, hypophosphatémie, hypoalbuminémie, hypomagnésiémie.
Affections psychiatriques
Très fréquent: troubles du sommeil (16,2%).
Fréquent: confusion, anxiété, dépression, modification de l'humeur.
Affections du système nerveux
Très fréquent: neuropathie périphérique sensitive (47,8%), vertiges (17,3%), tremblements (10,8%).
Fréquent: neuropathie périphérique, neuropathie périphérique sensitivomotrice, syncope, somnolence, léthargie, diminution du niveau de conscience, céphalées, dysgueusie, paresthésie.
Affections oculaires
Fréquent: vision trouble, cataracte.
Affections de l'oreille et du labyrinthe
Fréquent: vertiges.
Affections cardiaques
Fréquent: insuffisance cardiaque décompensée, fibrillation auriculaire, tachycardie.
Affections vasculaires
Fréquent: thrombose veineuse profonde, hypotension, hypertension.
Affections respiratoires, thoraciques et médiastinales
Très fréquent: toux (20,5%), dyspnée (20,2%).
Fréquent: embolie pulmonaire, toux productive, nez bouché, douleur oropharyngée, dysphonie, saignements de nez, dyspnée à l'effort.
Occasionnel: dyspnée, pneumopathie interstitielle y compris des cas de pneumopathie inflammatoire*.
Affections gastrointestinales
Très fréquent: constipation (36,7%), diarrhée (33,8%), nausées (17,6%), vomissements (11,5%).
Fréquent: douleurs abdominales, douleurs épigastriques, stomatite, sécheresse buccale, ventre ballonné.
Occasionnel: hémorragies gastro-intestinales*.
Affections hépatobiliaires
Fréquent: augmentation de l'alanine-aminotransférase.
Occasionnel: hyperbilirubinémie, élévation des paramètres hépatiques et hépatite*.
Affections de la peau et du tissu sous-cutané
Fréquent: éruption cutanée, prurit, peau sèche, hyperhydrose, sueurs nocturnes.
Très rares: syndrome de Stevens-Johnson*, nécrolyse épidermique toxique*, réaction médicamenteuse accompagnée d'une éosinophilie et de symptômes systémiques (DRESS)*.
Affections musculosquelettiques et systémiques
Très fréquent: dorsalgies (18,7%), faiblesse musculaire (13,7%), douleurs osseuses (13,6%), spasmes musculaires (13,0%).
Fréquent: arthralgie, douleurs de l'appareil locomoteur, douleurs musculo-squelettiques dans le thorax, douleurs dans une des extrémités/douleurs dans les membres.
Affections du rein et des voies urinaires
Fréquent: lésion rénale aiguë, insuffisance rénale, insuffisance rénale aiguë, affection rénale chronique, rétention urinaire, augmentation de la créatininémie.
Affections des organes de reproduction et du sein
Fréquent: douleurs pelviennes.
Troubles généraux et anomalies au site d'administration
Très fréquent: fatigue (37,1%), œdème périphérique (33,8%), fièvre (23,7%), asthénie (18,2%).
Fréquent: aggravation de l'état général, douleurs thoraciques, douleurs, frissons, douleurs thoraciques non cardiaques, œdème.
Occasionnel: syndrome de lyse tumorale*.
Investigations
Fréquent: perte de poids.
Lésions, intoxications et complications liées aux procédures
Fréquent: chute.
* = Données de pharmacovigilance
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Dans les études, le pomalidomide a été administré à des doses uniques allant jusqu'à 50 mg chez des volontaires sains et à des doses répétées de 10 mg une fois par jour chez des patients atteints d'un myélome multiple. Il n'a pas été rapporté d'événements indésirables graves liés à un surdosage. Le pomalidomide est éliminé par hémodialyse.
Propriétés/Effets
Code ATC
L04AX06
Mécanisme d'action
Le pomalidomide est un dérivé du thalidomide et se présente sous forme d'un racémate. Il possède des propriétés immunomodulatrices ainsi qu'anti-angiogénétiques.
Le pomalidomide inhibe la prolifération des cellules malignes hématopoïétiques et il inhibe également in vitro la prolifération des lignées cellulaires résistantes au lénalidomide. Il stimule la prolifération des lymphocytes T et la cytotoxicité médiée par les cellules tueuses naturelles (Natural killer - NK), il inhibe la production des cytokines pro-inflammatoires (par ex. TNF-α et IL-6) et bloque la migration et l'adhésion des cellules endothéliales.
Le pomalidomide se lie directement à la protéine cereblon (CRBN), qui fait partie d'un complexe E3-ligase comprenant les protéines DDB1 (DNA damage-binding protein 1), Cullin 4 (CUL4) et Roc1 et pouvant inhiber l'auto-ubiquitination du CRBN dans le complexe. Les E3-ubiquitines ligases sont responsables de la polyubiquitination de différentes protéines substrats et peuvent partiellement expliquer les effets pléiotropiques observés sur les cellules dans le cadre d'un traitement par le pomalidomide.
In vitro, en présence du pomalidomide, les protéines substrats Aiolos et Ikaros font l'objet d'une ubiquitination et ensuite d'une dégradation, avec pour résultat des effets cytotoxiques et immunomodulateurs. In vivo, le traitement par le pomalidomide donne lieu à une diminution du taux d'Ikaros chez les patients atteints d'un myélome multiple en rechute réfractaire au lénalidomide.
Pharmacodynamique
Voir la section sur le mécanisme d'action.
Efficacité clinique
Le pomalidomide en association avec le bortézomib et la dexaméthasone (PVd) à plus faible dose
Une étude clinique multicentrique, randomisée, ouverte, à 2 bras de phase III (CC-4047-MM-007) a évalué l'efficacité et la sécurité d'emploi du pomalidomide en association avec le bortézomib et la dexaméthasone (PVd) à plus faible dose comparé au bortézomib et à la dexaméthasone (Vd) à plus faible dose chez des patients adultes préalablement traités, qui avaient reçu au moins un traitement antérieur, dont un devait être un traitement par le lénalidomide.
Les patients dans le bras PVd ont reçu 4 mg de pomalidomide par voie orale aux jours 1 à 14 d'un cycle de 21 jours. La dexaméthasone à plus faible dose de 20 mg a été administrée une fois par jour aux jours 1, 2, 4, 5, 8, 9, 11 et 12 d'un cycle de 14 jours du cycle 1 jusqu'au cycle 8 inclus et ensuite une fois par jour aux jours 1, 2, 8 et 9 de chaque cycle suivant de 21 jours à partir du cycle 9. Les patients âgés de >75 ans ont reçu 10 mg de dexaméthasone conformément au même schéma thérapeutique que les patients plus jeunes. Le bortézomib (1,3 mg/m2/dose) a été administré aux jours 1, 4, 8 et 11 du cycle de 21 jours, du cycle 1 jusqu'au cycle 8 inclus et ensuite à la même dose aux jours 1 et 8 du cycle de 21 jours à partir du cycle 9. Dans le bras Vd, le bortézomib et la dexaméthasone ont été administrés à la même dose et conformément au même schéma thérapeutique que celui du bras PVd. Le traitement a été poursuivi jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable. Lorsqu'il était nécessaire de traiter les symptômes d'une toxicité, les doses avaient été réduites ou le traitement temporairement interrompu ou arrêté.
Au total, 559 patients ont été randomisés: 281 dans le bras PVd et 278 dans le bras Vd. 54% des patients étaient de sexe masculin d'un âge médian de la population totale de 68 ans (min., max.: 27, 89 ans). Environ 70% des patients étaient réfractaires au lénalidomide (71,2% dans le bras PVd, 68,7% dans le bras Vd). Dans l'ensemble, les caractéristiques démographiques et pathologiques étaient généralement bien équilibrées dans les deux bras.
Le critère d'efficacité principal était la survie sans progression (SSP). La SSP était définie comme le délai entre la randomisation et la progression de la maladie ou le décès. La réponse a été évaluée par un comité indépendant d'évaluation de la réponse (IRAC, Independent Response Adjudication Committee) conformément aux critères de réponse de l'International Myeloma Working Group (IMWG) en utilisant l'analyse de la population en intention de traiter comme analyse principale. Le critère d'évaluation secondaire était la survie globale.
Pour la population en ITT, après un suivi médian de 15,9 mois, la médiane de SSP était de 11,20 mois (IC à 95%: 9,66; 13,73) dans le bras PVd. Dans le bras Vd, la médiane de SSP était de 7,10 mois (IC à 95%: 5,88; 8,48). La SSP dans le bras PVd était significativement plus longue que dans le bras Vd: HR 0,61 (IC à 95%: 0,49; 0,77) et suggérait une réduction du risque de progression de la maladie ou de décès de 39% dans le bras PVd.
Chez les patients avec une CrCl initiale <45 ml/min, la médiane de SSP était de 7,16 mois (IC à 95%: 1,94; 10,74) dans le bras PVd (N = 37) comparé à 4,44 mois (IC à 95%: 2,83; 9,49) dans le bras Vd (N = 38); HR = 1,06, IC à 95%: 0,61; 1,83.
D'après une analyse intermédiaire actuelle de la survie globale (gel des données le 15 septembre 2018; suivi médian de 26,2 mois) 116/281 (41,3%) des patients dans le bras PVd étaient décédés et 126/278 (45,3%) dans le bras Vd. La médiane de la survie globale d'après l'évaluation de Kaplan-Meier était de 40,5 mois pour le bras PVd et 30,5 mois pour le bras Vd; HR = 0,91, IC à 95%: 0,70, 1,18, avec un taux global d'événements de 43,3%.
Le pomalidomide en association avec la dexaméthasone à faible dose (Pd)
Une étude de phase III (CC-4047-MM-003) et une étude de phase II (CC-4047-MM-002) ont été réalisées pour évaluer l'efficacité et la sécurité d'Imnovid.
La phase II
L'étude MM-002 a été réalisée chez 221 patients présentant un myélome multiple en rechute et réfractaire, qui étaient réfractaires au dernier traitement de leur myélome et qui avaient reçu le lénalidomide et le bortézomib.
Le pomalidomide 4 mg a été administré jusqu'à la progression de la maladie, seul pendant 21 jours d'un cycle de 28 jours ou en association avec de la dexaméthasone (40 mg par semaine, administrés aux jours 1, 8, 15 et 22 de chaque cycle de 28 jours).
Près de 81% des patients étaient réfractaires au lénalidomide, 74% au bortézomib et 64% aux lénalidomide et bortézomib (selon les critères de l'International Myeloma Working Group (IMWG)).
Pour le pomalidomide plus dexaméthasone, la survie médiane sans progression (critère d'évaluation principal) était de 16,6 semaines et pour le pomalidomide en monothérapie, elle était de 10,7 semaines. Le taux de réponse pour le pomalidomide plus dexaméthasone s'élevait à 30,1% contre 9,3% pour le pomalidomide en monothérapie.
La phase III
L'étude de phase III (MM-003) a comparé le traitement par pomalidomide plus dexaméthasone (Pom+LD-dex) avec une monothérapie par la dexaméthasone (HD-dex) chez 455 patients présentant un myélome multiple en rechute et réfractaire qui avaient reçu au moins deux traitements et chez qui tant le lénalidomide que le bortézomib avaient été un échec et dont la maladie avait évolué au cours du dernier traitement.
La médiane de SSP (critère d'évaluation principal) dans le bras POM+LD-dex était de 15,7 semaines (IC à 95%: 13,0-20,1) et de 8 semaines dans le bras HD-dex (IC à 95%: 7,0- 9,0) avec un rapport des risques instantanés (hazard ratio [HR]) de 0,45 (IC à 95%: 0,35-0,59); (p <0,001).
Dans le bras pomalidomide la survie globale médiane n'était pas encore obtenu. La différence entre les deux bras de traitement dans la survie globale était statistiquement significative (hazard ratio de 0,53 (IC à 95%: 0,37-0,74); p <0,001).
Pharmacocinétique
Absorption
Le pomalidomide atteint une concentration plasmatique maximale (Cmax) en 2 à 3 heures et après administration d'une dose orale unique, son absorption est >70%. La biodisponibilité est à peu près dose-proportionnelle. Sa biodisponibilité n'est pas affectée lorsqu'il est administré avec un repas riche en graisses et en calories. Le pomalidomide peut être administré en dehors des repas.
Distribution
Le volume de distribution à l'état d'équilibre est de 62 à 138 litres. La concentration dans le sperme de volontaires sains s'élevait à environ 67% de la concentration plasmatique. La liaison aux protéines est faible.
Métabolisme
Le pomalidomide est le principal composant retrouvé dans la circulation sanguine (environ 70% de la radioactivité plasmatique). Aucun métabolite n'est présent à une concentration supérieure à 10% de la radioactivité totale dans le plasma.
Les principales voies métaboliques de la radioactivité excrétée sont une hydroxylation suivie d'une glucuroconjugaison ainsi que d'une hydrolyse. In vitro, le CYP1A2 et le CYP3A4 ont été identifiés comme les principales enzymes impliquées dans l'hydroxylation du pomalidomide induite par le CYP, avec des contributions supplémentaires minimes du CYP2C19 et du CYP2D6.
La Cmax du pomalidomide avait augmenté de 14% tandis que l'ASC du pomalidomide avait diminué de 32% chez 14 volontaires sains de sexe masculin qui, après une dose orale unique de 4 mg de pomalidomide, avaient fumé 25 cigarettes par jour pendant une période de 10 jours, par rapport à 13 volontaires sains de sexe masculin non-fumeurs.
Élimination
La demi-vie plasmatique du pomalidomide est d'environ 9,5 heures chez les volontaires sains et d'environ 7,5 heures chez les patients atteints d'un myélome multiple. La clairance corporelle totale (Cl/F) moyenne est d'environ 7 à 10 l/heure.
73% de la dose sont éliminés dans les urines et 15% dans les fèces, environ 2% et 8% étant éliminés sous forme de pomalidomide dans les urines et les fèces.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Les paramètres pharmacocinétiques sont légèrement modifiés chez les patients présentant une insuffisance hépatique (définie selon les critères de Child-Pugh) par rapport aux volontaires sains. Par rapport aux volontaires sains, l'exposition moyenne au pomalidomide est augmentée de 51% (intervalle de confiance à 90% [9% à 110%] ) chez les patients présentant une insuffisance hépatique légère, de 58% (intervalle de confiance à 90% [13% à 119%]) chez les patients présentant une insuffisance hépatique modérée et de 72% (intervalle de confiance à 90%, [24% à 138%]) chez les patients présentant une insuffisance hépatique sévère.
Troubles de la fonction rénale
Les analyses pharmacocinétiques de population ont montré que les paramètres pharmacocinétiques du pomalidomide n'étaient pas significativement modifiés chez les patients présentant une insuffisance rénale (définie par la clairance de la créatinine ou le débit de filtration glomérulaire estimé [DFGe]) par rapport aux patients ayant une fonction rénale normale (ClCr ≥60 ml/minute). L'exposition systémique (ASC) normalisée moyenne au pomalidomide était de 98,2% avec un intervalle de confiance à 90% [77,4% à 120,6%] chez les patients atteints d'insuffisance rénale modérée (DFGe ≥30 et ≤45 ml/minute/1,73 m2) par rapport aux patients ayant une fonction rénale normale. L'exposition systémique (ASC) normalisée moyenne au pomalidomide était de 100,2% avec un intervalle de confiance à 90% [79,7% à 127,0%] chez les patients atteints d'insuffisance rénale sévère ne nécessitant pas de dialyse (ClCr <30 ml/minute ou DFGe <30 ml/minute/1,73 m2) par rapport aux patients ayant une fonction rénale normale. L'exposition systémique (ASC) normalisée moyenne au pomalidomide était augmentée de 35,8% avec un IC à 90% [7,5% à 70,0%] chez les patients atteints d'insuffisance rénale sévère nécessitant des dialyses (ClCr <30 ml/minute nécessitant des dialyses) par rapport aux patients ayant une fonction rénale normale.
Patients âgés
Chez des patients âgés de 61 à 82 ans, les paramètres pharmacocinétiques moyens de l'ASC (0-∞) et de la Cmax étaient en général similaires à ceux de patients plus jeunes.
Enfants et adolescents
On ne dispose pas de données sur la pharmacocinétique la population pédiatrique.
Données précliniques
Génotoxicité/carcinogénicité
Le pomalidomide n'a pas montré d'effets mutagènes dans les tests de mutation sur cellules bactériennes et cellules de mammifères et n'a pas induit d'aberrations chromosomiques dans des lymphocytes de sang périphérique humains ni de formation de micronoyaux dans les érythrocytes polychromatiques dans la moelle osseuse de rats ayant reçu des doses allant jusqu'à 2'000 mg/kg/jour. Il n'a pas été réalisé d'études de cancérogenèse.
Fertilité et développement embryonnaire précoce
Dans une étude de fertilité et de développement embryonnaire précoce chez le rat, le pomalidomide a été administré chez les mâles et les femelles aux doses de 25, 250 et 1'000 mg/kg/jour. L'examen de l'utérus le 13e jour de gestation a montré une diminution du nombre moyen d'embryons viables et une augmentation des pertes post-implantatoires à toutes les doses. Par conséquent, la DSENO pour ces effets était inférieure à 25 mg/kg/jour (ASC24h: 39'960 ng·h/ml; exposition 99 fois supérieure comparativement à la dose clinique la plus faible testée de 4 mg). Lorsque les mâles traités dans cette étude ont été accouplés avec les femelles non traitées, tous les paramètres utérins ont été comparables à ceux des témoins. Sur la base de ces résultats, les effets observés ont été imputés au traitement des femelles.
Développement embryonnaire et fœtal
Chez le rat et le lapin, le pomalidomide s'est avéré tératogène lorsqu'il a été administré pendant la phase d'organogenèse majeure. Dans l'étude de toxicité sur le développement embryofœtal chez le rat, des malformations ou une absence de vessie, absence de thyroïde et fusion et défaut d'alignement des vertèbres thoraciques et lombaires (corps vertébraux et/ou arcs neuraux) ont été observées à toutes les doses (25, 250 et 1'000 mg/kg/jour). Il n'a pas été mis en évidence de toxicité maternelle dans cette étude. Par conséquent, la DSENO maternelle a été de 1'000 mg/kg/jour et la DSENO en termes de toxicité sur le développement a été <25 mg/kg/jour (l'ASC24h était de 34'340 ng·h/ml le 17e jour de gestation à cette dose la plus faible testée et le rapport d'exposition était de 85 comparativement à une dose clinique de 4 mg). Chez le lapin, le pomalidomide administré à des doses allant de 10 à 250 mg/kg a induit des malformations embryonnaires et fœtales. Une augmentation des anomalies cardiaques a été observée à toutes les doses, avec une incidence significativement plus élevée à la dose de 250 mg/kg/jour. Aux doses de 100 et 250 mg/kg/jour, on a observé une légère augmentation des pertes post-implantatoires et une légère baisse du poids fœtal à la naissance. A la dose de 250 mg/kg/jour, les malformations fœtales consistaient en anomalies des membres (flexion et/ou rotation des membres antérieurs et/ou postérieurs, doigts non attachés ou absents) et malformations osseuses associées (absence d'ossification métacarpienne, défaut d'alignement des phalanges et métacarpes, doigt absent, absence d'ossification des phalanges et tibia court non ossifié ou courbé); dilatation modérée du ventricule latéral du cerveau; position anormale de l'artère sous-clavière droite; absence du lobe intermédiaire du poumon; implantation basse des reins; modifications de la morphologie hépatique; ossification absente ou incomplète du pelvis; augmentation du nombre moyen de côtes thoraciques surnuméraires et diminution du nombre moyen de tarses ossifiés. Une faible réduction de la prise de poids des mères, une diminution significative des triglycérides et une diminution significative des poids absolu et relatif de la rate ont été observées aux doses de 100 et 250 mg/kg/jour. La DSENO maternelle a été de 10 mg/kg/jour et la DSENO sur le développement a été inférieure 10 mg/kg/jour (l'ASC24h était de 418 ng·h/ml le 19e jour de gestation à cette dose la plus faible testée, soit une valeur similaire à celle obtenue avec une dose clinique de 4 mg).
Remarques particulières
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver dans l'emballage d'origine, ne pas conserver au-dessus de 25 °C et tenir hors de la portée des enfants.
Remarques concernant la manipulation
Comme c'est le cas pour les cytostatiques, la manipulation et l'élimination d'Imnovid nécessitent une prudence particulière (voir également la rubrique «Posologie/Mode d'emploi»).
Les femmes en âge de procréer qui manipulent le pomalidomide, doivent porter des gants de protection.
Les gélules ne doivent pas être ouvertes ou écrasées. Si la poudre de pomalidomide entre en contact avec la peau, laver immédiatement et abondamment la peau au savon et à l'eau. En cas de contact avec les muqueuses, rincer abondamment à l'eau.
Numéro d’autorisation
61249 (Swissmedic)
Titulaire de l’autorisation
Celgene GmbH, Zurich
Mise à jour de l’information
Janvier 2020
Reviews (0)
Recently Viewed




Syoss Hairspray Max Hold 400ml
Das Syoss Max Hold Haarspray sorgt während 48 Stunden für einen mega starken Halt, ist dabei leicht ..
16.67 CHF


Free consultation with an experienced pharmacist
Describe the symptoms or the right drug - we will help you choose its dosage or analogue, place an order with home delivery or just consult.
We are 14 pharmacists and 0 bots. We will always be in touch with you and will be able to communicate at any time.
Bestsellers

Milupa Aptamil Pepti Syneo Can 400g
Product code: 7748334Aptamil Pepti Syneo is a special food for babies from birth who are allergic to cow's milk protein a..
55.08 CHF


Weleda Baby Calendula Wind- und Wetterbalsam 30ml
Product code: 5455461Der Weleda Baby Calendula Wind- und Wetterbalsam schützt die sensible Babyhaut intensiv bei Kälte un..
15.35 CHF


HerpoTherm herpes pen
Product code: 7798882Herpotherm® - the heating pen Cold sores are not only unattractive but can also be very painful. Th..
78.48 CHF



