
Lucentis Inj Lös 1.65 mg / ml 0.165ml Fertspr 0.165
Lucentis Inj Lös 1.65 mg/0.165ml Fertspr 0.165 ml
-
1,215.90 CHF
- Price in reward points: 3131

- Availability: Not available
- Brand: NOVARTIS SCHWEIZ AG
- Product Code: 5907288
- ATC-code S01LA04
- EAN 7680632770019
Ingredients:
Histidin, Polysorbat 20, Trehalose-2-Wasser, Ranibizumab 1.65 mg, Histidin hydrochlorid-1-Wasser.

Description
 Deutsch
Deutsch French
French Italian
ItalianZusammensetzung
Wirkstoffe
Ranibizumabum.
Hilfsstoffe
α,α-trehalosum dihydricum, Histidinum, Histidinum hydrochloricum monohydricum, Polysorbatum 20, Aqua ad iniect.
Darreichungsform und Wirkstoffmenge pro Einheit
Durchstechflasche: Ranibizumab 10 mg/ml (Durchstechflasche zu 2.3 mg Ranibizumab in 0.23 ml Lösung).
Fertigspritze: Ranibizumab 10 mg/ml (Fertigspritze zu 1.65 mg Ranibizumab in 0.165 ml Lösung).
Indikationen/Anwendungsmöglichkeiten
Lucentis ist indiziert bei Erwachsenen für die Behandlung:
- der exsudativen (feuchten) altersbezogenen Makuladegeneration (feuchte AMD).
- eines Visusverlustes durch ein Diabetisches Makulaödem (DME).
- der mässig schweren bis schweren nicht-proliferativen diabetischen Retinopathie (NPDR) bzw. proliferativen diabetischen Retinopathie (PDR),
- eines Visusverlustes durch ein Makulaödem infolge eines retinalen Venenverschlusses (retinaler Venenastverschluss BRVO und retinaler Zentralvenenverschluss CRVO).
- einer aktiven, den Visus beeinträchtigenden choroidalen Neovaskularisation (CNV).
- eines Visusverlustes durch choroidale Neovaskularisation (CNV) infolge einer pathologischen Myopie (PM).
Lucentis ist indiziert bei Frühgeborenen für die Behandlung der Frühgeborenen Retinopathie (RPM):
- RPM in Zone I (Krankheitsstadium 1+, 2+, 3 oder 3+), Zone II (Krankheitsstadium 3+) oder AP-RPM (aggressive posteriore RPM).
Dosierung/Anwendung
Lucentis darf nur durch einen qualifizierten Ophthalmologen angewendet werden, der über eine adäquate Infrastruktur verfügt. Lucentis wird in den Glaskörper (intravitreal) injiziert. Eine Lucentis Durchstechflasche (Erwachsene und Frühgeborene) enthält 0.23 ml, eine Fertigspritze (nur für Erwachsene) enthält 0.165 ml: Beide sind zum einmaligen Gebrauch für einen Patienten bestimmt.
Übliche Dosierung
Dosierung für Erwachsene
Die empfohlene Dosis von 0.5 mg wird als intravitreale Einzelinjektion appliziert. Dies entspricht einem Injektionsvolumen von 0.05 ml. Der Abstand zwischen zwei Injektionen in dasselbe Auge darf nicht kürzer als 1 Monat sein.
Die Behandlung wird mit einer Injektion monatlich begonnen, bis der maximale Visus erreicht ist und/oder keine Anzeichen von Krankheitsaktivität mehr vorhanden sind. Bei Patienten mit feuchter AMD, DME, mässig schwerer bis schwerer NPDR oder PDR, und BRVO oder CRVO können initial drei oder mehr aufeinanderfolgende monatliche Injektionen notwendig sein.
Danach sollten die Abstände zwischen den Kontrolluntersuchungen und die Behandlungsintervalle vom Arzt festgelegt werden. Bei den Kontrolluntersuchungen wird die Krankheitsaktivität durch klinische Untersuchung, durch Kontrolle des Visus und/oder durch bildgebende Verfahren (z.B. optische Kohärenztomographie und/oder Fluoreszein-Angiographie) beurteilt.
Die Kontroll- und Behandlungsintervalle können gemäss der vorliegenden Krankheitsaktivität und des beobachteten Therapieverlaufs schrittweise verlängert oder verkürzt werden.
Falls aufgrund der ärztlichen Beurteilung angezeigt, kann Lucentis auch monatlich an einem gegebenen Auge angewendet werden.
Die Behandlung eines Visusverlustes durch CNV muss individuell für jeden Patienten auf Grundlage der Krankheitsaktivität festgelegt werden. Für CNV infolge von einer pathologischen Myopie (PM) oder für andere, nicht im Rahmen einer AMD auftretende CNV, liegen keine Erfahrungen über einen Behandlungszeitraum von mehr als einem Jahr vor.
Dosierung für Frühgeborene
Die empfohlene Dosis für Lucentis bei Frühgeborenen beträgt 0.1 mg, die als einmalige intravitreale Injektion appliziert wird. Dies entspricht einem Injektionsvolumen von 0.01 ml. Bei Frühgeborenen wird die Behandlung der Frühgeborenen-Retinopathie (ROP) mit einer Einzeldosis eingeleitet, die an beiden Augen am selben Tag verabreicht werden kann. Insgesamt können bei Anzeichen von Krankheitsaktivität bis zu drei Injektionen pro Auge innerhalb von sechs Monaten verabreicht werden. Wenn mehr als eine Injektion erforderlich ist, sollte der zeitliche Abstand zwischen zwei Injektionen mindestens einen Monat betragen.
Patienten mit Leberfunktionsstörungen
Es liegen keine Untersuchungen vor. Da die systemisch verfügbare Menge vernachlässigbar ist, sind keine speziellen Vorsichtsmassnahmen vorgesehen.
Patienten mit Nierenfunktionsstörungen
Für Patienten mit eingeschränkter Nierenfunktion ist keine Dosisanpassung notwendig (s. «Pharmakokinetik»).
Ältere Patienten (ab 65 Jahren)
Es ist keine Dosisanpassung notwendig.
Kinder und Jugendliche
Lucentis wird für den Gebrauch bei Kindern und Jugendlichen nicht empfohlen, da nicht ausreichend Daten zur Sicherheit und Wirksamkeit in dieser Patientengruppe vorliegen. Zu jugendlichen Patienten im Alter von 12 bis 17 Jahren mit Visusverlust durch CNV liegen begrenzte Daten vor. (s. «Eigenschaften/Wirkungen»).
Art der Anwendung
Vor der intravitrealen Verabreichung sollte eine gründliche Anamnese hinsichtlich möglicher Überempfindlichkeitsreaktionen erhoben werden (s. «Warnhinweise und Vorsichtsmassnahmen»).
Die Verabreichung von Lucentis muss unter aseptischen Bedingungen (geeignete Räumlichkeiten, keimfreie Abdeckung, Handschuhe, Utensilien) erfolgen. Geeignete Anästhesie und ein topisches Breitband-Mikrobizid zur Desinfektion der periokulären Haut, des Lids und der okulären Oberfläche sollten vor Durchführung der Injektion angewendet werden.
Der gesamte Inhalt der Durchstechflasche wird mit einer Spritze und einer 5 µm Filternadel entnommen. Vor der intravitrealen Injektion wird die Filternadel entfernt und die Injektionsnadel (13 mm, 30 Gauge) auf die Spritze aufgesetzt. Der Inhalt soll soweit ausgestossen werden, dass die Kolbenspitze der Spritze auf der Markierung für die 0.05 ml (50 µl) liegt. Der Inhalt einer Durchstechflasche ist zur Verabreichung einer Einzeldosis bestimmt. Die nicht gebrauchte Lösung ist zu verwerfen.
Bei Erwachsenenen sollte die Injektionsnadel 3.5-4.0 mm hinter dem Limbus vollständig eingestochen werden. Dabei ist die horizontale Mittellinie zu vermeiden und die Nadel zum Zentrum des Augapfels zu richten.
Das Injektionsvolumen wird langsam abgegeben, und bei Folgeinjektionen ist auf einen Wechsel der Injektionsstelle auf der Sklera zu achten.
Nach der Injektion muss der Augeninnendruck des Patienten überwacht werden. Die Überwachung sollte die Kontrolle der Durchblutung der Sehnervenpapille unmittelbar nach der Injektion, Augeninnendruckmessung innert 30 Minuten, Ophthalmoskopie, Spaltlampenuntersuchung sowie Funduskontrolle nach 2-7 Tagen umfassen. Der Patient muss instruiert werden, allfällige Zeichen einer Endophthalmitis sofort dem Arzt zu melden (s. «Warnhinweise und Vorsichtsmassnahmen»).
Bei Frühgeborenen muss die Injektionsnadel 1.0 bis 2.0 mm hinter dem Limbus eingeführt werden, wobei die Nadel in Richtung des Sehnervs zeigt. Dann wird das Injektionsvolumen von 0.01 ml verabreicht.
Kontraindikationen
Überempfindlichkeit gegenüber Ranibizumab oder einem der Hilfsstoffe.
Lucentis ist kontraindiziert bei Patienten mit Infektionen im oder um das Auge, sowie bei Patienten mit aktiver intraokulärer Entzündung.
Warnhinweise und Vorsichtsmassnahmen
Reaktionen durch intravitreale Injektionen
Bei einer intravitrealen Injektion kann es zu infektiösen Endophthalmitiden und Netzhautablösungen kommen. Bei der Injektion von Lucentis sind aseptische Injektionstechniken anzuwenden. Zudem sollten die Patienten während der auf die Injektion folgenden Tage beobachtet werden, um eine Infektion frühzeitig zu erkennen und zu behandeln (s. «Unerwünschte Wirkungen»).
Bei Erwachsenen wurde ein vorübergehender Anstieg des Augeninnendrucks innerhalb von 60 Minuten nach der Injektion von Lucentis beobachtet. Es wurde auch über langanhaltenden erhöhten intraokulären Druck berichtet. Sowohl der intraokuläre Druck als auch die Perfusion der zentralen Retinalarterie müssen überwacht und entsprechend behandelt werden (s. «Unerwünschte Wirkungen»).
Beidseitige Behandlung
Die Sicherheit und Wirksamkeit einer gleichzeitigen Behandlung mit Lucentis an beiden Augen wurde nicht in dafür angelegten Studien geprüft.
Die begrenzt vorliegenden Daten zur bilateralen Anwendung von Lucentis (einschliesslich einer Verabreichung am selben Tag) weisen, verglichen mit einer unilateralen Behandlung, nicht auf ein erhöhtes Risiko für systemische unerwünschte Ereignisse hin.
Arterielle thromboembolische Ereignisse
Es besteht ein potentielles Risiko für arterielle thromboembolische Ereignisse bei der intravitrealen Applikation von VEGF (vascular endothelial growth factor)-Inhibitoren. Bei Patienten mit einem bekannten Risiko für Schlaganfälle (die z.B. bereits einen Schlaganfall oder eine transiente ischämische Attacke erlitten hatten) ist das Risiko möglicherweise erhöht.
Immunogenität
In allen Behandlungsgruppen zeigten 0-3% der noch nicht behandelten Patienten eine Immunreaktion gegenüber Lucentis. Bei monatlicher Applikation wurden nach 12-24 Monaten schwache Antikörper-Titer in 1-6% der Patienten beobachtet. Diese Immunogenitäts-Daten spiegeln den Prozentsatz der Patienten, bei denen ein Elektrochemilumineszenz-Test positive Resultate ergab. Die Daten waren stark abhängig von der Sensitivität und der Spezifität des Tests. Die klinische Signifikanz der Immunreaktion auf Lucentis ist zur Zeit unklar. Bei einigen Patienten mit den höchsten Immunoreaktivitätstitern wurden Iritis und Viritis beobachtet.
Patientengruppen mit eingeschränkt verfügbaren Daten
Die Anwendung von Lucentis ist bisher nicht bei Patienten mit aktiver systemischer Infektion oder mit weiteren Augenerkrankungen wie Netzhautablösung oder Makulaloch untersucht worden.
Lucentis und Laserphotokoagulation
Sowohl die Anwendung von Lucentis bei Patienten, welche sich in der Vorgeschichte einer Laserphotokoagulation unterzogen haben als auch eine gleichzeitige Anwendung von Lucentis und Laserphotokoagulation wurden untersucht. Falls Lucentis am selben Tag wie eine Laserphotokoagulation gegeben werden sollte, darf die Injektion frühestens 30 Minuten nach der Laserphotokoagulation erfolgen.
Dauer der Behandlung
Für CNV infolge einer pathologischen Myopie (PM) oder für andere, nicht im Rahmen einer AMD auftretende CNV, liegen keine Erfahrungen über einen Behandlungszeitraum von mehr als einem Jahr vor.
Interaktionen
Es wurden keine speziellen Interaktionsstudien durchgeführt.
Schwangerschaft/Stillzeit
Schwangerschaft
Es gibt keine Daten zur Anwendung von Ranibizumab bei schwangeren Frauen.
Studien mit Cynomolgus-Affen ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädigende Wirkungen in Bezug auf Schwangerschaft oder embryonale/fötale Entwicklung (s. «Präklinische Daten»). Ranibizumab hemmt den VEGF-A, einen wichtigen angiogenen Faktor für die Bildung von neuen Blutgefässen während der embryonalen und fötalen Entwicklung und Plazentabildung. Die systemische Exposition von Ranibizumab ist nach der okulären Anwendung gering. Wegen des Wirkungsmechanismus muss Ranibizumab als potentiell teratogen und embryo-/fetotoxisch angesehen werden und darf während der Schwangerschaft nicht angewendet werden, es sei denn es ist klar notwendig. Bei Patientinnen, die schwanger werden möchten, sollte Ranibizumab 3 Monate vor der Empfängnis abgesetzt werden.
Frauen im gebärfähigen Alter
Gebärfähige Frauen sollten während und bis 30 Tage nach Behandlung wirksame Methoden zur Schwangerschaftsverhütung anwenden.
Stillzeit
Es ist nicht bekannt, ob Lucentis in die Muttermilch ausgeschieden wird. Da zahlreiche Substanzen in die Muttermilch ausgeschieden werden, und die Möglichkeit einer Resorption und Beeinträchtigung des Wachstums und der Entwicklung des Kindes besteht, wird empfohlen, während der Behandlung mit Lucentis nicht zu stillen.
Fertilität
Der Einfluss von Lucentis auf die männliche und weibliche Fertilität wurde nicht untersucht.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Die Behandlung mit Lucentis kann vorübergehende Sehstörungen verursachen, wodurch die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen beeinflusst werden können (s. «Unerwünschte Wirkungen»). Patienten, die solche Anzeichen verspüren, dürfen nicht Auto fahren oder Maschinen bedienen, bis diese vorübergehenden Sehstörungen abgeklungen sind.
Unerwünschte Wirkungen
Aus den drei klinischen Studien der Phase III zur Behandlung der feuchten AMD bilden insgesamt 1'315 Patienten die Basis für die Sicherheitsüberlegungen. Alle Patienten wurden mindestens 24 Monate mit Lucentis behandelt, mit der empfohlenen Dosis von 0.5 mg wurden 440 Patienten therapiert.
Schwerwiegende unerwünschte Ereignisse im Zusammenhang mit dem Injektionsvorgang waren: Endophthalmitis, rhegmatogene Netzhautablösung, Einriss der Retina und iatrogene traumatische Katarakt (s. «Warnhinweise und Vorsichtsmassnahmen»).
Bei mit Lucentis behandelten Patienten wurden auch intraokuläre Entzündung und erhöhter Augeninnendruck beobachtet (s. «Warnhinweise und Vorsichtsmassnahmen»).
Bei Patienten mit feuchter AMD, die in den kontrollierten Phase-III-Studien zur feuchten AMD FVF2598g (MARINA), FVF2587g (ANCHOR) und FVF3192g (PIER) mit 0.5 mg Lucentis behandelt wurden, traten die unten aufgelisteten unerwünschten Ereignisse häufiger auf (mindestens 2 Prozentpunkte) als bei Patienten der Kontrollgruppen (Scheininjektion (s. «Eigenschaften/Wirkungen») oder PDT mit Verteporfin). Deshalb wurden sie als potenzielle unerwünschte Arzneimittelwirkungen (UAWs) beschrieben. Die unten aufgeführten Daten zur Sicherheit beinhalten darüber hinaus alle unerwünschten Ereignisse von denen angenommen wird, dass sie zumindest potenziell durch die Injektion als solche oder durch das Arzneimittel verursacht werden und bei den 440 Patienten auftraten, die mit 0.5 mg Lucentis gegen feuchte AMD in der Kombinationstherapie behandelt wurden.
Patienten Kollektiv mit DR
Die Sicherheit von Lucentis wurde in einer 24-monatigen klinischen Studie in der Protocol-S-Studie untersucht, die u.a. 191 mit Ranibizumab behandelte Patienten mit diabetischer Retinopathie (DR) einschloss. Die beobachteten okulären und nicht-okulären Ereignisse standen im Einklang mit dem, was bei einer diabetischen Patientenpopulation mit DR zu erwarten wäre, oder sie ähnelten im Hinblick auf ihre Häufigkeit und Schwere den Ereignissen, die in früheren klinischen Studien zu Lucentis beobachtet wurden.
Patienten Kollektiv CNV
Die Sicherheit von Lucentis wurde in einer 12-monatigen klinischen Studie (MINERVA) untersucht, an der 171 mit Ranibizumab behandelte Patienten mit Visusverlust durch CNV teilnahmen (s. «Eigenschaften/Wirkungen»). Das Sicherheitsprofil bei diesen Patienten war konsistent mit jenem früherer klinischer Studien mit Lucentis.
Patienten Kollektiv CNV infolge von PM
Die Sicherheit von Lucentis wurde in der 12-monatigen klinischen Studie RADIANCE untersucht. An dieser Studie nahmen 224 Patienten mit CNV infolge von PM teil, die mit Ranibizumab behandelt wurden (s. «Eigenschaften/Wirkungen»). Häufigkeit und Schwere der in dieser Studie aufgetretenen okulären und nicht-okulären Ereignisse waren mit denen in den Studien bei feuchter AMD vergleichbar.
Population mit Frühgeborenen Retinopathie (RPM)
- Die Sicherheit von Lucentis 0.1 mg wurde in einer sechsmonatigen klinischen Studie (RAINBOW) untersucht, die 77 mit Ranibizumab behandelte Frühgeborene mit RPM umfasste (s. «Eigenschaften/Wirkungen»). Die okulären Nebenwirkungen, die in der RAINBOW-Studie beobachtet wurden, waren konsistent mit jenen, die auch bei Erwachsenen unter der Behandlung mit Ranibizumab 0.5 mg auftraten oder die ein Ereignis im Rahmen der RPM darstellen. Die nicht-okulären Nebenwirkungen in dieser klinischen Studie entsprachen generell denen, die bei diesen Patienten mit multiplen Komorbiditäten aufgrund der Frühgeburt zu erwarten wären. Aus theoretischen Gründen muss in Betracht gezogen werden, dass die Reifung anderer Organe nach einer Behandlung mit anti-VEGFs verzögert werden könnte, und darum Komplikationen auftreten könnten.
Häufigkeiten: «Sehr häufig» (>1/10), «häufig» (>1/100 <1/10), «gelegentlich» (>1/1'000 <1/100), «selten» (>1/10'000 <1/1'000), «sehr selten» (<1/10'000).
Infektionen und parasitäre Erkrankungen
Sehr häufig: Nasopharyngitis (12.5-16.4%).
Häufig: Influenza, Harnwegsinfektionen.
Erkrankungen des Blutes und des Lymphsystems
Häufig: Anämie.
Erkrankungen des Immunsystems
Häufig: Hypersensitivitätsreaktionen.
Psychiatrische Erkrankungen
Häufig: Angstzustände.
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen (8-15%).
Häufig: Schlaganfall.
Augenerkrankungen
Sehr häufig: Intraokuläre Entzündungen (10-18%), Glaskörperentzündung (2.3-10%), Glaskörperabhebung (18-19%), Netzhautblutungen (25%), Sehstörungen (6.6-10.5%), Augenschmerzen (27-32%), Mouches volantes (7-25%), Bindehautblutung (55-72%), Augenirritation (12-15%), Fremdkörpergefühl im Auge (12-15%), verstärkter Tränenfluss (10-14%), Blepharitis (8-11%), Pruritus (8.5-10%).
Häufig: Retina-Degeneration, Störungen der Retina, Retina-Abhebung, Risse der Retina, Abhebung des retinalen Pigmentepithels, Risse im retinalen Pigmentepithel, Seh-Verschlechterung, Glaskörperblutungen und –Störungen, Uveitis, Iritis, Iridocyclitis, (subkapsuläre) Katarakt, posteriore Kapselsack-Trübung, Keratitis punctata, Kornea-Abrasionen, Trübungen des Kammerwassers, verschwommenes Sehen, Blutungen an der Injektionsstelle, Augenblutungen, (allergische) Konjunktivitis, Ausscheidungen am Auge, Photopsie, Photophobie, Augenbeschwerden, Schmerzen und Ödeme des Augenlids, Hyperämie der Konjunktiva.
Gelegentlich: Endophthalmitis, Hypopyon, Hyphaema, Keratopathie, Verklebung der Iris, Kornea-Einschmelzungen und –Ödeme, Streifen (Striae) der Kornea, Schmerzen und Irritationen an der Einstichstelle, Erblindung, Irritationen des Augenlids.
Herzerkrankungen, Gefässerkrankungen
Arterielle thromboembolische Ereignisse nach Definition der Antiplatelet Trialists' Collaboration (1994) wie gefässbedingte Todesfälle, nicht-fatale Myokardinfarkte, nicht-fatale ischämische Schlaganfälle und nicht-fatale hämorrhagische Schlaganfälle wurden mit der systemischen Verfügbarkeit von hochpotenten VEGF-Inhibitoren in Zusammenhang gebracht. Im ersten Jahr betrug der Anteil der thromboembolischen Ereignisse in den beiden mit Lucentis behandelten Patientengruppen (0.3 und 0.5 mg) 2.3%; im Vergleich dazu waren es in der Kontrollgruppe nur 1.3%. Im 2. Jahr der Studie MARINA waren es in beiden Behandlungsgruppen 3.0%, während dem es in der Kontrollgruppe 3.2% waren.
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Häufig: Husten.
Erkrankungen des Gastrointestinaltrakts
Häufig: Nausea.
Erkrankungen der Haut und des Unterhautgewebes
Gelegentlich: Allergische Reaktionen (Ausschlag, Urticaria, Pruritus, Erythema).
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Sehr häufig: Gelenkschmerzen (8-12%).
Untersuchungen
Erhöhter Augeninnendruck.
Immunogenität
Wie bei allen therapeutischen Proteinen besteht die Möglichkeit einer Immunantwort bei Patienten, die mit Lucentis behandelt wurden. Die Immunogenitätsdaten spiegeln den Prozentsatz von Patienten wieder, die in Immunoassays positiv auf Antikörper gegen Ranibizumab getestet wurden. Die Daten waren stark abhängig von der Sensitivität und der Spezifität der Assays.
In den AMD Studien zeigten in allen Behandlungsgruppen 0% bis 3% der noch nicht behandelten Patienten eine Immunreaktion gegenüber Lucentis. Nach der monatlichen Verabreichung von Lucentis über 12 bis 24 Monate wurden die Antikörper gegen Ranibizumab bei ungefähr 1% bis 8% der Patienten mit neovaskulärer AMD nachgewiesen.
In den DME Studien zeigten in allen Behandlungsgruppen 0% bis 2% der noch nicht behandelten Patienten eine Immunreaktion gegenüber Lucentis. Nach der monatlichen Verabreichung von Lucentis über 12 Monate wurden die Antikörper gegen Ranibizumab bei ungefähr 2% bis 4% der Patienten mit DME detektiert.
In den RVO Studien zeigten in allen Behandlungsgruppen 2% bis 3% der noch nicht behandelten Patienten eine Immunreaktion gegenüber Lucentis. Nach der monatlichen Verabreichung von Lucentis über 12 Monate wurden die Antikörper gegen Ranibizumab bei ungefähr 4% bis 5% der Patienten mit RVO detektiert.
Die klinische Signifikanz der Immunoreaktivität auf Lucentis ist zur Zeit unklar.
In den gepoolten Daten der abgeschlossenen, randomisierten, doppelblinden, klinischen Studien traten bei DME Patienten nicht-schwerwiegende, nicht- okuläre Wundinfektionen/-entzündungen unter 0.5 mg Ranibizumab häufiger auf als unter den Kontrollbehandlungen (1.85/100 Patientenjahre vs. 0.27/100 Patientenjahre). Ein Zusammenhang mit Ranibizumab ist bislang nicht bekannt. Es besteht jedoch das theoretische Risiko eines Zusammenhangs dieser Ereignisse mit der VEGF-Inhibition.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
Überdosierung
Aus den klinischen Studien zur feuchten AMD und aus Post-Marketing-Beobachtungen wurden Fälle von unbeabsichtigter Überdosierung (Injektion von grösseren Volumina als den empfohlenen 0.05 ml Lucentis) berichtet.
Anzeichen und Symptome
Die dabei am häufigsten auftretenden unerwünschten Wirkungen sind erhöhter Augeninnendruck und Augenschmerzen.
Behandlung
Falls eine zu hohe Dosis verabreicht wurde, sollte der Augeninnendruck überwacht und je nach Einschätzung durch den behandelnden Arzt gegebenenfalls behandelt werden.
In klinischen Studien erhielten Patienten mit feuchter AMD und DME Dosierungen von bis zu 2 mg Ranibizumab in einem Injektionsvolumen von 0.05 ml bis 0.10 ml. Art und Häufigkeit der unerwünschten okulären und systemischen Ereignisse entsprachen den für die Dosis 0.5 mg (in 0.05 ml) Lucentis berichteten.
Eigenschaften/Wirkungen
ATC-Code
S01LA04
Wirkungsmechanismus
Der Wirkstoff von Lucentis (Ranibizumab) ist ein humanisiertes, rekombinantes monoklonales Antikörperfragment (Fab) gegen den humanen vaskulären endothelialen Wachstumsfaktor A (VEGF-A). Es bindet mit hoher Affinität an VEGF-A und an dessen Isoformen. Die Isoformen, wie z.B. VEGF121 und VEGF165 entstehen durch alternatives mRNA-Spleissen, die Isoform VEGF110 durch Proteolyse. Die Bindung von Ranibizumab an VEGF-A und dessen Isoformen inhibiert die Aktivierung der Rezeptoren VEGFR-1 und VEGFR-2 auf der Oberfläche der Endothelzellen.
Pharmakodynamik
Die Aktivierung der Rezeptoren VEGFR-1 und -2 führt zur Proliferation von Endothelzellen, zur Neovaskularisation und zum Flüssigkeitsaustritt aus den Gefässen. Es wird angenommen, dass alle diese Faktoren zur Progression der neovaskulären Form der altersbedingten Makuladegeneration (AMD), der Entwicklung von CNV, inklusive CNV infolge einer pathologischen Myopie (PM), von diabetischen Makulaödemen (DME) und von retinalen Venenverschlüssen, die zu Visusverlust führen (RVO), beitragen.
Klinische Wirksamkeit
Behandlung der feuchten AMD
Die klinische Wirksamkeit und Sicherheit von Ranibizumab zur Behandlung der feuchten AMD wurde in drei randomisierten, doppelt-maskierten Studien bei insgesamt 1'323 Patienten (Lucentis: N=879, Kontrollgruppen N=444) mit neovaskulärer AMD untersucht. Als Kontrollarm diente in der Studie MARINA Scheinbehandlungen und in der Studie ANCHOR eine aktive Kontrolle mittels PDT mit Visudyne. Eingeschlossen wurden Patienten mit Läsionen in der Grösse von bis zu 12 Papillenflächen und einer Sehschärfe von 20/40 bis 20/320 (nach Snellen) im Studienauge. Das Durchschnittsalter der Patienten lag bei 77 Jahren. In den klinischen Studien wurden die Patienten angewiesen, sich selbstständig antimikrobielle Augentropfen zu applizieren (viermal täglich, jeweils 3 Tage vor und nach jeder Injektion).
In die 24 monatige Studie MARINA wurden 716 Patienten mit minimaler klassischer CNV oder okkulter CNV eingeschlossen. Sie erhielten monatliche intravitreale Injektionen von Lucentis 0.3 mg (N=238), Lucentis 0.5 mg (N=240) oder Scheininjektionen (N=238). Während der 24-monatigen Behandlungsphase erhielten die Patienten durchschnittlich 22 von 24 möglichen Behandlungen. Die Resultate der MARINA Studie nach 12 Monaten Behandlung wurden bei 90% der Patienten auch nach 24 Monaten Behandlung (1x/Monat) im Wesentlichen bestätigt.
In die 24 monatige Studie ANCHOR wurden 423 Patienten mit vorwiegend klassischen CNV-Läsionen eingeschlossen. Sie erhielten monatliche intravitreale Injektionen von Lucentis 0.3 mg und Schein-PDT (N=143), intravitreale Injektionen von Lucentis 0.5 mg und Schein-PDT (N=140) oder intravitreale Scheininjektionen und aktive Verteporfin-PDT (N=143). Die erste Schein- bzw. aktive Verteporfin-PDT wurde gemeinsam mit der anfänglichen Lucentis-Injektion appliziert. Danach erfolgte die Behandlung im Abstand von 3 Monaten, wenn am Studienauge weiterhin oder erneut Flüssigkeit aus den Gefässen austrat (Fluoreszein-Angiographie).
Die Resultate sind in den nachfolgenden Tabellen zusammengefasst:
Tabelle 0-1 Studie MARINA: Resultate nach 12 und 24 Monaten
Veränderung der Sehschärfe (in Buchstaben, ETDRS) | Monat | Scheinbehandlung | Ranibizumab 0.5 mg | Differenz |
|---|---|---|---|---|
Sehschärfe-Verlust <15 Buchstaben (%)b | Monat 12 | 62% | 95% | 32% |
Monat 24 | 53% | 90% | 37% | |
Sehschärfe-Gewinn ≥15 Buchstaben (%)b | Monat 12 | 5% | 34% | 29% |
Monat 24 | 4% | 33% | 29% | |
Durchschnittliche Veränderung der Sehschärfeb (Buchstaben) | Monat 12 | -10.5 (16.6) | +7.2 (14.4) | 17.5 |
Monat 24 | -14.9 (18.7) | +6.6 (16.5) | 21.1 | |
a nach Stratifizierung b p <0.01 | ||||
Tabelle 0-2 Studie ANCHOR: Resultate nach 12 und 24 Monaten
Veränderung der Sehschärfe | Verteporfin PDT | Ranibizumab 0.5 mg | Differenz | |
|---|---|---|---|---|
Sehschärfe-Verlust <15 Buchstaben (%)b | Monat 12 | 64% | 96% | 33% |
Monat 24 | 66% | 90% | 25% | |
Sehschärfe-Gewinn ≥15 Buchstaben (%)b | Monat 12 | 6% | 40% | 35% |
Monat 24 | 6% | 41% | 35% | |
Durchschnittliche Veränderung der Sehschärfeb (Buchstaben) | Monat 12 | -9.5 (16.4) | +11.3 (14.6) | 21.1 |
Monat 24 | -9.8 (16.4) | +10.7 (16.5) | 20.7 | |
a nach Stratifizierung b p <0.01 | ||||
In den beiden Studien MARINA und ANCHOR resultierte die unter der Behandlung mit 0.5 mg Lucentis nach Monat 12 beobachtete Verbesserung des Visus in einem Nutzen für den Patienten, gemessen anhand der drei Subskalen des National Eye Institute Visual Function Questionnaire (VFQ-25), die zuvor als sekundäre Endpunkte für die Wirksamkeit festgelegt worden waren (Tätigkeiten mit Bezug auf Nah- und Weitsehen sowie weitere, vom Sehen abhängige Tätigkeiten). Alle Unterschiede zwischen Lucentis 0.5 mg und den zwei Kontrollgruppen waren statistisch signifikant und klinisch relevant, mit p-Werten zwischen 0.009 bis <0.0001.
In die Studie PIER wurden 184 Patienten mit CNV-Läsionen (mit und ohne klassische Anteile) eingeschlossen. Sie erhielten während der ersten 3 Monate je eine monatliche intravitreale Injektion von Lucentis 0.3 mg bzw. von Lucentis 0.5 mg oder intravitreale Scheininjektion. Weitere Injektionen von Lucentis erfolgten im Abstand von 3 Monaten. Nach Monat 14 konnten diejenigen Patienten, welche eine Scheininjektion erhielten, ebenfalls mit Lucentis behandelt werden und ab dem Monat 19 waren häufigere Injektionen von Lucentis möglich. Die mit Lucentis behandelten Patienten in PIER erhielten durchschnittlich 10 Behandlungen innert 24 Monaten.
Der primäre Wirksamkeits-Endpunkt war die mittlere Veränderung der Sehschärfe während der 12 Monate. Nach einem anfänglichen Anstieg in der Zeit der monatlichen Injektionen, verloren die Patienten in der Phase der 3-monatlichen Injektionen an Sehschärfe, die nach 12 Monaten zum Ausgangspunkt zurückkehrte und dieser Effekt wurde in den meisten mit Lucentis behandelnden Patienten (82%) bei Monat 24 aufrechterhalten. Die Daten von einer begrenzten Anzahl derjenigen Patienten, welche nach über einem Jahr Scheinbehandlung zu einer Behandlung mit Lucentis gewechselt haben, deuten darauf hin, dass ein früher Therapiebeginn mit besserer Erhaltung der Sehschärfe assoziiert ist.
Tabelle 0-3 Studie PIER: Resultate nach 12 Monaten
Veränderung der Sehschärfe | Scheinbehandlung (n=63) | Ranibizumab 0.5 mg | Differenz |
|---|---|---|---|
Sehschärfe-Verlust <15 Buchstaben (%)b | 49 | 90 | 37 |
Sehschärfe-Gewinn ≥15 Buchstaben (%)b | 10 | 13 | 2 |
Durchschnittliche Veränderung der Sehschärfeb | -16.3 (22.3) | -0.2 (13.1) | 14.7 |
a nach Stratifizierung b p <0.0001 | |||
Die Phase-IIIb Studie SAILOR wurde bei Behandlungsnaïven, wie auch vorbehandelten Patienten mit CNV infolge von AMD durchgeführt. SAILOR war eine 1-Jahres Multizenterische Studie. Das primäre Ziel der Studie war die Abschätzung der Inzidenz von okulären und nicht-okulären unerwünschten Wirkungen während der 12 Behandlungsmonate. 2378 Patienten wurden in 2 Gruppen randomisiert und erhielten während 3 Monaten 0.3 mg bzw. 0.5 mg Lucentis jeden Monat appliziert; anschliessend wurde je nach Befund weiterbehandelt, mit Abständen von mindestens 1 Monat.
Insgesamt bestand kein Ungleichgewicht zwischen den beiden Gruppen in Bezug auf okuläre und nicht-okuläre unerwünschte Wirkungen. Es gab insbesondere auch kein Ungleichgewicht zwischen den beiden Gruppen in Bezug auf die Zahl der Schlaganfälle. Unter 0.3 mg waren es 8/1'169 Patienten (0.7%, 95%CI: 0.3% bis 1.3%). Unter 0.5 mg waren es 15/1'209 Patienten (1.2%, 95% CI: 0.7% bis 2%). Patienten mit bekannten Risiko-Faktoren wie z.B. ein vorangegangener Schlaganfall oder eine vorangegangene transiente ischämische Attacke, haben vermutlich ein erhöhtes Risiko für einen Schlaganfall während der Behandlung mit Lucentis.
Behandlung des Visusverlustes durch DME
Die klinische Wirksamkeit und Sicherheit von Lucentis bei Patienten mit Visusverlust durch ein Diabetisches Makulaödem (DME) wurde in der Studie RESTORE bei insgesamt 345 Patienten mit Visusverlust durch DME untersucht. Die Studie hatte 3 Arme: In Arm 1 wurde Patienten (N=116) initial Ranibizumab 0.5 mg intravitreal als Monotherapie injiziert und eine Schein-Laserphotokoagulation durchgeführt. In Arm 2 (N=118) wurde initial Ranibizumab 0.5 mg intravitreal injiziert und eine Laserphotokoagulation durchgeführt. In Arm 3 (N=111) wurde eine initiale Monotherapie mit Laserphotokoagulation und eine Scheininjektion durchgeführt.
Die Behandlung mit Ranibizumab wurde mit monatlichen intravitrealen Injektionen fortgesetzt und unterbrochen, wenn sich der Visus des Patienten unter Lucentis an drei aufeinanderfolgenden Untersuchungsterminen stabilisiert hatte. Nach einer Unterbrechung wurde die Behandlung wieder aufgenommen, wenn sich der Visus des Patienten durch Fortschreiten des DME verschlechtert hatte. Wiederbehandlungen mit Laserphotokoagulation wurden am gleichen Tag, mindestens 30 Minuten vor der Ranibizumab Injektion, gemäss der ETDRS-Kriterien durchgeführt.
Die Resultate sind in den folgenden Tabellen zusammengefasst:
Tabelle 0-4 Studie RESTORE: Resultate nach 12 Monaten
Veränderung der bestmöglich korrigierten Sehschärfe | Ranibizumab | Ranibizumab | Laser |
|---|---|---|---|
Durchschnittliche Veränderung des best korrigierten Visus vom 1. zum 12. Monat in Buchstaben, verglichen mit dem Visus zu Beginn der Studie (Standardabweichung)a | 6.1 (6.4) | 5.9 (7.9) | 0.8 (8.6) |
Durchschnittliche Veränderung des best korrigierten Visus im 12. Monat in Buchstaben, verglichen mit dem Visus zu Beginn der Studie (Standardabweichung) | 6.8 (8.3)a | 6.4 (11.8)b | 0.9 (11.4) |
Zunahme des best korrigierten Visus ≥10 Buchstaben (% der Patienten) | 37.4c | 43.2 | 15.5 |
Zunahme des best korrigierten Visus ≥15 Buchstaben (% der Patienten) | 22.6d | 22.9e | 8.2 |
a p <0.0001, b p=0.0004, c p=0.0001, d p=0.0032, e p=0.0021 | |||
Bei der RESTORE-Erweiterungsstudie handelte es sich um eine offene, multizentrische, 24-monatige Erweiterungsstudie. 240 Patienten, die die 12-monatige Kernstudie abgeschlossen hatten, traten in die Erweiterungsstudie ein und wurden nach Bedarf (PRN: pro re nata) im selben Auge, das in der Kernstudie als Studienauge definiert worden war, mit 0.5 mg Ranibizumab behandelt. Die Behandlung wurde nach einem Abfall der BCVA aufgrund von DME monatlich bis zum Erreichen einer stabilen BCVA verabreicht. Ausserdem erfolgte eine Laserbehandlung gemäss den ETDRS-Leitlinien, falls dies vom Prüfarzt als notwendig erachtet wurde.
Die durchschnittliche Anzahl von Ranibizumab-Injektionen bei Patienten, die in der Kernstudie mit Ranibizumab behandelt worden waren, betrug in der 24-monatigen Erweiterungsphase 6.4. Von den 74 Patienten aus dem Laserbehandlungsarm der Kernstudie erhielten 59 (79%) Patienten zu irgendeinem Zeitpunkt während der Erweiterungsphase Ranibizumab. Die durchschnittliche Anzahl von Ranibizumab-Injektionen während der 24-monatigen Erweiterungsphase betrug bei diesen 59 Patienten 8.1 Injektionen. Die Anteile der Patienten, die während der Erweiterungsphase keine Behandlung mit Ranibizumab benötigten, betrugen in den Gruppen, die zuvor eine Behandlung mit Ranibizumab, Ranibizumab + Laser bzw. Laser allein erhalten hatten, 19%, 25% bzw. 20%.
Die Ergebnisse sind in der folgenden Tabelle zusammengefasst:
Tabelle 0-5 Resultate nach 36 Monaten in der RESTORE-Erweiterungsstudie
Ergebnisparameter gegenüber Baseline in der Kernstudie | Vorher Ranibizumab | Vorher Ranibizumab | Vorher Laser |
Mittlere Veränderung der BCVA gegenüber Baseline in der Kernstudie nach 36 Monaten (SD) | 8.0 (10.09) | 6.7 ( 9.59) | 6.0 ( 9.35) |
Gewinn von ≥10 Buchstaben gegenüber Baseline in der Kernstudie bzw. BCVA ≥84 (%) nach 36 Monaten | 39 (47.0) | 37 (44.6) | 31 (41.9) |
Gewinn von ≥15 Buchstaben gegenüber Baseline in der Kernstudie bzw. BCVA ≥84 (%) nach 36 Monaten | 23 (27.7) | 25 (30.1) | 16 (21.6) |
n = Anzahl der Patienten, für die Werte sowohl bei Baseline (Kernstudie, Monat 0) als auch beim Besuch in Monat 36 verfügbar waren. * 59 (79%) der 74 Patienten mit vorheriger Laserbehandlung erhielten in der Erweiterungsstudie Ranibizumab. | |||
Das in dieser 24-monatigen Erweiterungsstudie beobachtete Langzeitsicherheitsprofil von Ranibizumab deckt sich mit dem bekannten Sicherheitsprofil von Lucentis.
In der Phase-IIIb-Studie RETAIN wurden 372 Patienten mit Visusverlust durch DME randomisiert folgenden intravitrealen Injektionen zugeteilt:
- Ranibizumab 0.5 mg mit gleichzeitiger Laserphotokoagulation im Rahmen eines «Treat and Extend»(TE)-Schemas (n=121),
- Ranibizumab 0.5 mg als Monotherapie im Rahmen eines TE-Schemas (n = 128) oder
- Ranibizumab 0.5 mg als Monotherapie im Rahmen eines PRN-Schemas (n = 123).
- In allen Gruppen wurde die Ranibizumab-Behandlung mit monatlichen intravitrealen Injektionen eingeleitet und bis zur Erreichung einer stabilen BCVA bei mindestens drei aufeinanderfolgenden monatlichen Untersuchungen fortgeführt. Die Laserphotokoagulation erfolgte bei Baseline am selben Tag wie die erste Ranibizumab-Injektion und anschliessend nach Bedarf gemäss den ETDRS-Kriterien. Im Rahmen des TE-Schemas wurde Ranibizumab im weiteren Verlauf in Abständen von 2- oder maximal 3 Monaten verabreicht. Beim PRN-Schema wurde die BCVA monatlich beurteilt, und Ranibizumab wurde dann, falls erforderlich, beim selben Besuch verabreicht. In allen Gruppen wurde die monatliche Behandlung nach einem Abfall der BCVA aufgrund einer DME-Progression wieder aufgenommen und bis zum erneuten Erreichen einer stabilen BCVA fortgeführt. Die Studiendauer betrug 24 Monate.
- In der RETAIN-Studie betrug die Zahl der Injektionen im Durchschnitt (Median) 12.4 (12.0) bei der TE-Ranibizumab + Laser-Behandlungsgruppe, 12.8 (12.0) bei der TE-Ranibizumab-Monotherapie- Behandlungsgruppe und 10.7 (10.0) bei der PRN-Ranibizumab-Behandlungsgruppe. Die Anzahl der benötigten, planmässigen Behandlungstermine war nach 3 initialen monatlichen Behandlungsterminen 13 unter dem TE-Schema, verglichen mit 20 monatlichen Terminen, die unter dem PRN-Schema benötigt wurden. Unter beiden Behandlungsschemata hielten mehr als 70% der Patienten ihre BCVA bei einer Visitenfrequenz von ≥2 Monaten aufrecht. Zusätzliche Laserbehandlungen waren unter dem entsprechenden TE-Schema nicht mit einer geringeren durchschnittlichen Zahl von Ranibizumab-Injektionen assoziiert.
Die Ergebnisse sind in der folgenden Tabelle zusammengefasst:
Tabelle 0-6 Resultate in der RETAIN-Studie
Ergebnisparameter gegenüber Baseline | TE Ranibizumab | TE Ranibizumab | PRN Ranibizumab |
|---|---|---|---|
Mittlere durchschnittliche Veränderung der BCVA von Monat 1 bis Monat 12 (SD) | 5.9 (5.5)b | 6.1 (5.7)b | 6.2 (6.0) |
Mittlere durchschnittliche Veränderung der BCVA von Monat 1 bis Monat 24 (SD) | 6.8 (6.0) | 6.6 (7.1) | 7.0 (6.4) |
Mittlere Veränderung der BCVA nach 24 Monaten (SD) | 8.3 (8.1) | 6.5 (10.9) | 8.1 (8.5) |
Gewinn von ≥10 Buchstaben bzw. BCVA ≥84 (%) nach 24 Monaten | 43.6 | 40.8 | 45.3 |
Gewinn von ≥15 Buchstaben bzw. BCVA ≥84 (%) nach 24 Monaten | 25.6 | 28.0 | 30.8 |
Verlust von ≥10 Buchstaben nach 24 Monaten | 2.6 | 7.2 | 3.4 |
Verlust von ≥15 Buchstaben nach 24 Monaten | 0.9 | 4.0 | 2.6 |
b p <0.0001 | |||
In den DME-Studien ging die BCVA-Verbesserung in allen Behandlungsgruppen mit einer allmählichen Verringerung der mittleren CRT einher.
Die Behandlung der mässig schweren bis schweren nicht-proliferativen diabetischen Retinopathie (NPDR) bzw. proliferativen diabetischen Retinopathie (PDR)
Die klinische Sicherheit und Wirksamkeit von Lucentis bei Patienten mit proliferativer diabetischer Retinopathie (PDR) wurde in eine multizentrische, randomisierte, aktiv kontrollierte Nichtunterlegenheitsstudie der Phase III im Parallel-Design (Protocol S), an der 305 Patienten (394 Studienaugen) mit PDR mit oder ohne DME (diabetisches Makulaödem) bei Baseline teilnahmen und in der 0.5 mg intravitreal injiziertes Ranibizumab mit der Standardbehandlung mit panretinale Photokoagulation (PRP) verglichen wurde, beurteilt. Insgesamt wurden 191 Augen (48.5%) randomisiert und der Behandlung mit 0.5 mg Ranibizumab und 203 Augen wurden (51.5%) randomisiert und der Behandlung mit PRP zugewiesen. Insgesamt 88 Augen (22.3%) wiesen bei Baseline ein DME auf: 42 (22.0%) und 46 (22.7%) der Augen in der Ranibizumab- bzw. der PRP-Gruppe. Insgesamt 306 Augen (77.7%) wiesen bei Baseline kein DME auf: 149 (78.0%) und 157 (77.3%) der Augen in der Ranibizumab- bzw. der PRP-Gruppe.
Nach 2 Jahren Behandlung hatte sich der BCVA (best corrected visual acuity) Score in der Lucentis-Gruppe von der Baseline um +2.7 Buchstaben und in der PRP Gruppe von der Baseline um -0.7 Buchstaben verändert. Die Differenz von 3.5 Buchstaben lag innerhalb der non-inferiority margin, sodass die Non-Inferiorität von Lucentis versus PRP bestätigt wurde.
Die Änderung des Schweregrads der diabetischen Retinopathie wurde anhand von Fundusfotos unter Verwendung des Schweregrad-Scores für diabetische Retinopathie (DRSS) aus der frühen Studie zur diabetischen Retinopathie (ETDRS) beurteilt. In dieser Studie war bei 41.8% der Augen unter der Behandlung mit Ranibizumab (n=189) in Monat 12 eine Verbesserung beim DRSS um mindestens 2 Stufen eingetreten, verglichen mit 14.6% bei den mit PRP behandelten Augen (n=199).
In einer Metaanalyse von 3 randomisierten, doppelblinden, aktiv kontrollierten Phase-III-Studien [D2301 (RESTORE), D2303 (REVEAL) und D2305 (REFINE)], durchgeführt in insgesamt 875 Patienten mit DME, zeigten 48,4% der 315 mit Ranibizumab behandelten Patienten mit mittelschwerer bis schwerer NPDR oder PDR (n = 192) eine Verbesserung der DRSS um mindestens 2 Stufen im 12. Monat, verglichen mit 14,6% der Laserbehandelte Patienten (n = 123).
Behandlung des Visusverlustes durch ein Makulaödem infolge RVO
Die klinische Sicherheit und Wirksamkeit von Lucentis bei Patienten mit Visusverlust durch ein Makulaödem infolge eines retinalen Venenverschlusses (RVO) wurde in zwei randomisierten, doppelt-maskierten, kontrollierten Studien BRAVO (N=397) und CRUISE (N=392) untersucht. In beiden Studien erhielten die Patienten entweder 0.3 mg Ranibizumab oder 0.5 mg Ranibizumab oder eine Scheinbehandlung. In BRAVO, war eine Laserphotokoagulation als Rettungsbehandlung zu jedem Zeitpunkt während der Studie ab dem 3. Monat in allen Studienarmen erlaubt. Die Resultate aus BRAVO und CRUISE sind in folgenden Tabellen zusammengestellt.
Tabelle 0-7 Studie BRAVO: Resultate bei Monat 6 und 12
Veränderung der Sehschärfe | Scheinbehandlung | Ranibizumab 0.5 mg |
|---|---|---|
Durchschnittliche Veränderung der best-korrigierten Sehschärfe bei Monat 6 in Buchstaben, verglichen mit der Sehschärfe zu Beginn der Studiea | +7.3 | +18.3 |
Durchschnittliche Veränderung der best-korrigierten Sehschärfe bei Monat 12 in Buchstaben, verglichen mit der Sehschärfe zu Beginn der Studie | +12.1 | +18.3 |
Anteil der Patienten mit Sehschärfe-Gewinn ≥15 Buchstaben (%) bei Monat 6 | 28.8% | 61.1% |
Anteil der Patienten mit Sehschärfe-Gewinn ≥15 Buchstaben (%) bei Monat 12 | 43.9% | 60.3% |
Anteil der Patienten mit Laserrescue über 12 Monaten | 61.4% | 34.4% |
a p<0.0001 | ||
Tabelle 0-8 Studie CRUISE: Resultate bei Monat 6 und 12
Veränderung der Sehschärfe | Scheinbehandlung | Ranibizumab 0.5 mg |
|---|---|---|
Durchschnittliche Veränderung der best-korrigierten Sehschärfe bei Monat 6 in Buchstaben, verglichen mit der Sehschärfe zu Beginn der Studiea | +0.8 | +14.9 |
Durchschnittliche Veränderung der best-korrigierten Sehschärfe bei Monat 12 in Buchstaben, verglichen mit der Sehschärfe zu Beginn der Studie | +7.3 | +13.9 |
Anteil der Patienten mit Sehschärfe-Gewinn ≥15 Buchstaben (%) bei Monat 6 | 16.9% | 47.7% |
Anteil der Patienten mit Sehschärfe-Gewinn ≥15 Buchstaben (%) bei Monat 12 | 33.1% | 50.8% |
a p<0.0001 | ||
Mit Ranibizumab behandelte Patienten zeigten in beiden Studien (BRAVO und CRUISE) eine kontinuierliche Verringerung der zentralen Netzhautdicke.
Neben der Visusverbesserung nach Behandlung mit Ranibizumab bei Monat 6 und 12 auch die Lebensqualität der Patienten anhand des National Eye Institute Visual Function Questionnaire (VFQ-25) erhoben. Die Unterschiede zwischen der Ranibizumab 0.5 mg Gruppe und der Kontrollgruppe wurden bei Monat 6 mit p-Werten zwischen 0.02 und 0.0002 ermittelt.
Die Resultate der HORIZON Verlängerungsstudie zu BRAVO und CRUISE zeigten nach 12 Monaten folgendes:
Die reduzierte Behandlungsfrequenz in der Studie HORIZON hatte wenig Auswirkung auf BRVO-Patienten, welche ihre initiale Visusverbesserung, wie sie in der Studie BRAVO beobachtet wurde (+17.5 Buchstaben nach 24 Monaten mit einer Dosis von 0.5 mg und durchschnittlich 2.4 Injektionen im zweiten Jahr) behielten.
Hingegen zeigte sich die reduzierte Behandlungsfrequenz bei CRVO-Patienten mit einer Verringerung der in der Studie CRUISE gewonnenen Sehschärfe (+12 Buchstaben nach 24 Monaten mit einer Dosis von 0.5 mg und durchschnittlich 3.8 Injektionen im zweiten Jahr).
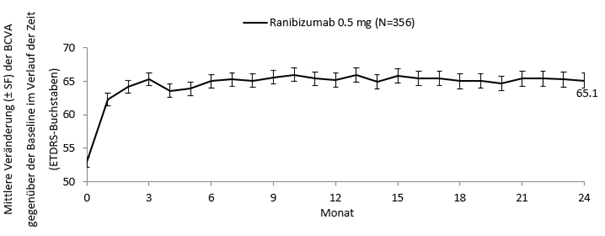
Studie E2401 (CRYSTAL) und Studie E2402 (BRIGHTER)
In den Studien BRIGHTER und CRYSTAL wurden die klinische Sicherheit und Wirksamkeit von Lucentis über 24 Monate bei Patienten mit Sehstörungen wegen eines RVO-bedingten Makulaödems bewertet. In die Studien wurden Patienten mit BRVO (n = 455) bzw. CRVO (n = 357) rekrutiert. In beiden Studien erhielten die Patienten 0.5 mg Ranibizumab nach Bedarf. BRIGHTER war eine 3-armige, randomisierte, aktiv kontrollierte Studie, in der 0.5 mg Ranibizumab als Monotherapie mit Ranibizumab in Kombination mit Laserphotokoagulation und mit Laserphotokoagulation alleine verglichen wurden. Nach 6 Monaten konnten die Teilnehmer im Arm mit der Laser-Monotherapie 0.5 mg Ranibizumab erhalten. CRYSTAL war eine einarmige Studie mit einer Monotherapie mit 0.5 mg Ranibizumab.
Die wichtigsten funktionellen und anatomischen Ergebnisse der BRIGHTER- und der CRYSTAL-Studie sind in Tabelle 0-9 und in den Abbildungen 1-0 und 2-0 dargestellt.
Tabelle 0-9 Ergebnisse in Monat 6 (BRIGHTER) und Monat 24 (BRIGHTER und CRYSTAL)
BRIGHTER | CRYSTAL | |||
|---|---|---|---|---|
Lucentis 0.5 mg | Lucentis 0.5 mg | Laser* | Lucentis 0.5 mg | |
Mittlere Veränderung der BCVA in Monat 6b (Buchstaben) (SD) | +14.8 | +14.8 | +6.0 | +12.0 |
Mittlere Veränderung der BCVA in Monat 24b (Buchstaben) (SD) | +15.5 | +17.3 | +11.6 | +12.1 |
Anteil der Patienten, die bei der BCVA im Monat 24 ≥15 Buchstaben erreichten | 52.8% | 59.6% | 43.3% | 49.2% |
Mittlere Anzahl an Injektionen (SD) (Monate 0-23) | 11.4 | 11.3 | N/A | 13.1 |
* Ab Monat 6 war eine Behandlung mit 0.5 mg Ranibizumab zulässig (24 Patienten wurden nur mit Laser behandelt). b p <0.0001 für beide Vergleiche in der BRIGHTER-Studie in Monat 6: Lucentis 0.5 mg gegenüber Laser und Lucentis 0.5 mg + Laser gegenüber Laser. | ||||
Abbildung 1-0 BRIGHTER: Mittlere Veränderung der BCVA gegenüber der Baseline über einen Zeitraum von 24 Monaten
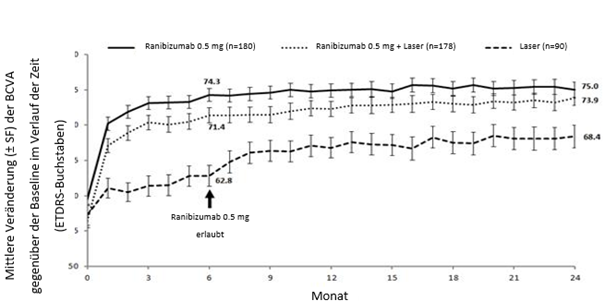
Abbildung 2-0 CRYSTAL: Mittlere Veränderung der BCVA gegenüber der Baseline über einen Zeitraum von 24 Monaten
Die Verbesserung des Visus fiel bei Patienten mit oder ohne Netzhautischämie ähnlich aus: In der BRIGHTER-Studie wiesen Patienten mit Netzhautischämie (n = 87) oder ohne Netzhautischämie (n = 35), die mit einer Ranibizumab-Monotherapie behandelt wurden, in Monat 24 eine mittlere Veränderung gegenüber der Baseline von +15.4 bzw. +12.9 Buchstaben auf. In der CRYSTAL-Studie wiesen Patienten mit Netzhautischämie (n = 107) oder ohne Netzhautischämie (n = 109) eine mittlere Veränderung gegenüber der Baseline von +11.1 bzw. +12.9 Buchstaben auf.
Bei Patienten mit einer Krankheitsdauer von <3 Monaten war in der BRIGHTER- bzw. CRYSTAL-Studie in Monat 1 eine Verbesserung der Sehschärfe von 13.3 bzw. 10.0 Buchstaben und in Monat 24 von 17.7 bzw. 13.2 Buchstaben zu beobachten. Bei einer Krankheitsdauer von ≥12 Monaten waren es in Monat 1 4.8 bzw. 7.9 Buchstaben, in Monat 24 waren es 8.4 bzw. 8.6 Buchstaben. Eine Initiierung der Behandlung bei Diagnosestellung ist zu erwägen.
Das Sicherheitsprofil von Ranibizumab, das in diesen 24-monatigen Studien beobachtet wurde, entspricht dem aus früheren Studien bekannten Sicherheitsprofil von Lucentis.
Behandlung eines Visusverlustes durch CNV – Studie G2301 (MINERVA)
Die klinische Sicherheit und Wirksamkeit von Lucentis bei Patienten mit Visusverlust durch CNV infolge von anderen Ätiologien als neovaskulärer AMD und PM wurden auf Grundlage der 12-Monats-Daten der randomisierten, doppelt-maskierten, Scheinbehandlungs-kontrollierten Studie G2301 (MINERVA) beurteilt. Für die Analyse wurden fünf Untergruppen nach Ätiologie vordefiniert (angioide Streifen, postentzündliche Retinochoroidopathie, zentrale seröse Chorioretinopathie, idiopathische Chorioretinopathie und andere Ätiologien). 178 Patienten wurden im Verhältnis 2:1 in einen der folgenden Arme randomisiert:
Ranibizumab 0.5 mg zu Beginn der Studie, gefolgt von einem individuellen Dosierungsschema je nach Krankheitsaktivität.
Scheininjektion zu Beginn der Studie, gefolgt von einem individuellen Dosierungsschema je nach Krankheitsaktivität.
Ab Monat 2 erhielten alle Patienten eine Behandlung mit Ranibizumab nach Bedarf. Primärendpunkt war die Veränderung der besten korrigierten Sehschärfe (BCVA) vom Beginn der Studie bis Monat 2.
Die wichtigsten Ergebnisse von MINERVA sind in Tabellen 0-10 und 0-11 sowie in Abbildung 3-0 zusammengestellt.
Tabelle 0-10 Resultate nach 2 Monaten (MINERVA)
Ranibizumab 0.5 mg | Scheinbehandlung | |
|---|---|---|
Mittlere BCVA-Veränderung vom Beginn der Studie bis Monat 2 (Buchstaben) (Least-Squares-Mittelwert) a | +9.5 | -0.4 |
Anteil Patienten mit einem Gewinn von ≥15 Buchstaben seit Beginn der Studie oder einem Wert von 84 Buchstaben in Monat 2 | 31.4% | 12.3% |
Anteil Patienten, die vom Beginn der Studie bis Monat 2 nicht mehr als 15 Buchstaben verloren | 99.2% | 94.7% |
Verringerung der mittleren Foveadicke vom Beginn der Studie bis Monat 2 (Least-Squares-Mittelwert) a | 77 µm | -9.8 µm |
a Einseitiger Vergleich (p <0.001) mit Scheinbehandlungskontrolle | ||
Abbildung 3-0: Mittlere Veränderung der BCVA vom Beginn der Studie bis Monat 12 im Zeitverlauf (MINERVA)
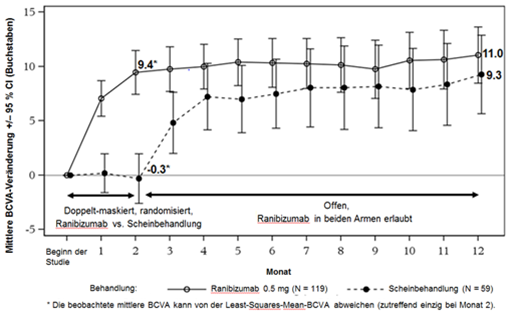
Beim Vergleich von Ranibizumab mit den Scheininjektionen in Monat 2 wurde eine konsistente Wirkung beobachtet, dies sowohl in der gesamten Studienpopulation, als auch in den einzelnen, gemäss der Ätiologie definierten Untergruppen.
Tabelle 0-11 Gesamtwirkung der Behandlung und Wirkung der Behandlung in den zu Beginn der Studie gemäss der Ätiologie definierten Untergruppen für die primäre Variable in Monat 2 (MINERVA)
Insgesamt und gegliedert nach Ätiologie bei Studienbeginn | Behandlungswirkung im Vergleich zur Scheinbehandlung (Buchstaben) | Patientenzahlen |
|---|---|---|
Gesamt | 9.9 | 175* |
Angioide Streifen | 14.6 | 27 |
Postentzündliche Retinochoroidopathie | 6.5 | 27 |
Zentrale seröse Chorioretinopathie | 5.0 | 23 |
Idiopathische Chorioretinopathie | 11.4 | 62 |
Sonstige Ätiologiena | 10.6 | 36 |
a CNV-Ätiologien, die nicht unter die anderen Untergruppen fallen * Anzahl Patienten mit für die Analyse verfügbaren Daten | ||
Die Verbesserung des Visus wurde von einer Verringerung der mittleren Foveadicke über einen Zeitraum von 12 Monaten begleitet.
Die mittlere Anzahl der Ranibizumab-Injektionen über einen Zeitraum von 12 Monaten in das Studienauge betrug im Ranibizumab-Arm 5.8 und in der Gruppe mit Scheininjektionen 5.4.
Kinder und Jugendliche
Fünf jugendliche Patienten im Alter von 12 bis 17 Jahren mit Visusverlust infolge CNV (1x subfoveale CNV bei einer Drusenpapille; je 1x juxtafoveale CNV und subfoveale CNV bei idiopathischer CNV; 2x subfoveale CNV bei Best disease) erhielten eine initiale Behandlung mit Ranibizumab 0.5 mg gefolgt von einem individualisierten Behandlungsschema auf Grundlage von Anzeichen der Krankheitsaktivität (z.B. Beeinträchtigung der Sehschärfe, intra-/subretinale Flüssigkeit, Blutungen oder Leckagen). Die BCVA-Veränderung vom Beginn der Studie bis Monat 12 verbesserte sich bei allen fünf Patienten und reichte von +5 bis +38 Buchstaben. Die Verbesserung des Visus wurde von einer Stabilisierung oder Verringerung der mittleren Foveadicke über einen Zeitraum von 12 Monaten begleitet (ΔCSFT0-12 Mte.: -286 μm bis +10 μm). Während der 12 Monate wurden 2 bis 5 Injektionen ins Studienauge verabreicht (s. «Dosierung/Anwendung»).
Behandlung eines Visusverlustes durch CNV infolge von PM
Die klinische Sicherheit und Wirksamkeit von Lucentis bei Patienten mit Visusverlust durch CNV infolge von PM wurden ausgehend von 12-Monats-Daten aus der randomisierten, doppelt maskierten, kontrollierten Pivotstudie RADIANCE beurteilt. Diese Studie war dazu ausgelegt, zwei verschiedene Dosierungsschemata von 0.5 mg Ranibizumab per intravitrealer Injektion im Vergleich zu Verteporfin PDT (vPDT, photodynamische Therapie mit Visudyne) zu evaluieren.
Die 277 Patienten wurden in einen der folgenden Arme randomisiert:
Gruppe I (0.5 mg Ranibizumab, Dosierungsplan beruhte auf Stabilitäts-Kriterien, definiert als keine Veränderung der bestkorrigierten Sehschärfe (BCVA) im Vergleich zu zwei vorhergehenden monatlichen Untersuchungen)
Gruppe II (0.5 mg Ranibizumab, Dosierungsplan beruhte auf Krankheitsaktivitäts-Kriterien, definiert als Visusverlust aufgrund von intra- oder subretinaler Flüssigkeit oder aktivem Flüssigkeitsaustritt infolge der CNV-Läsion mit Nachweis per OCT und/oder FA).
Gruppe III (vPDT - die Patienten konnten ab Monat 3 mit Ranibizumab behandelt werden)
Im Lauf der 12 Studienmonate erhielten die Patienten in Gruppe I durchschnittlich 4.6 Injektionen (zwischen 1 und 11 Injektionen) und in Gruppe II durchschnittlich 3.5 Injektionen (zwischen 1 und 12 Injektionen). In Gruppe II (in welcher Patienten die empfohlene Behandlung basierend auf Krankheitsaktivität erhielten, s. «Dosierung/Anwendung»), benötigten 50.9% der Patienten eine oder zwei Injektionen, 34.5% der Patienten drei bis fünf Injektionen und 14.7% der Patienten 6 bis 12 Injektionen innerhalb der 12-monatigen Studienperiode. 62.9% der Gruppe II-Patienten benötigten in den letzten sechs Monaten der Studie keine weiteren Injektionen.
Die wichtigsten Behandlungsergebnisse der Studie RADIANCE sind in Tabelle 0-12 und Abbildung 0-4 zusammengefasst.
Tabelle 0-12 Behandlungsergebnis bei Monat 3 und Monat 12 (RADIANCE)
Gruppe I | Gruppe II | Gruppe III | |
|---|---|---|---|
Monat 3 | |||
Mittlere durchschnittliche Veränderung der BCVA von Monat 1 bis Monat 3 gegenüber Baselinea (Buchstaben) | +10.5 | +10.6 | +2.2 |
Anteil der Patienten mit Verbesserung der BCVA um | |||
≥10 Buchstaben oder einer BCVA von insgesamt ≥84 Buchstaben | 61.9% | 65.5% | 27.3% |
≥15 Buchstaben oder einer BCVA von insgesamt ≥84 Buchstaben | 38.1% | 43.1% | 14.5% |
Monat 12 | |||
Anzahl der Injektionen bis Monat 12: | |||
Durchschnitt | 4.6 | 3.5 | N/A |
Medianwert | 4.0 | 2.0 | N/A |
Mittlere durchschnittliche Veränderung der BCVA von Monat 1 bis Monat 12 gegenüber Baseline (Buchstaben) | +12.8 | 12.5 | N/A |
Anteil der Patienten mit Verbesserung der BCVA um | |||
≥10 Buchstaben oder einer BCVA von insgesamt ≥84 Buchstaben | 69.5% | 69.0% | N/A |
≥15 Buchstaben oder einer BCVA von insgesamt ≥84 Buchstaben | 53.3% | 51.7 | N/A |
* Vergleichende Kontrolle bis Monat 3. Die in die vPDT-Gruppe randomisierten Patienten konnten ab Monat 3 eine Behandlung mit Ranibizumab erhalten (in Gruppe III erhielten 38 Patienten Ranibizumab ab Monat 3): p<0.00001 für den Vergleich mit der vPDT-Kontrolle | |||
Abbildung 0-4 Mittlere Veränderung der BCVA gegenüber Baseline BCVA im Zeitverlauf bis Monat 12 (RADIANCE)
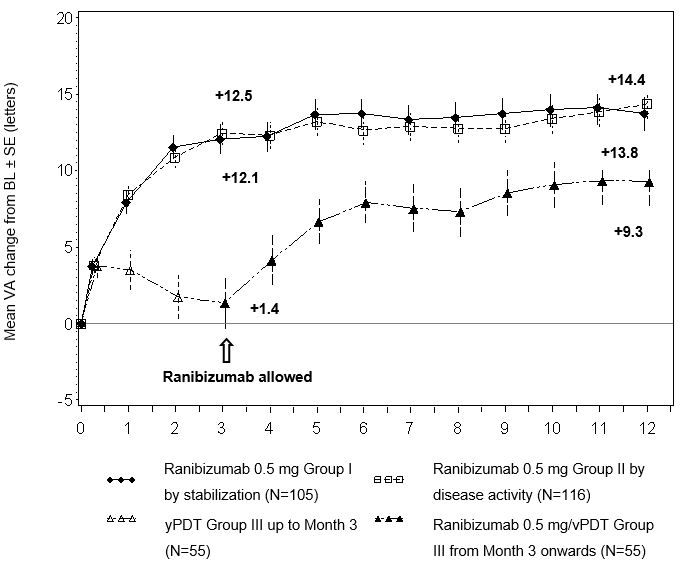
BL = Baseline; SE = Standardfehler des Mittelwerts.
Abbildung:
Mittlere Verbesserung der Sehschärfe gegenüber BL ± SE (Buchstaben)
Gruppe I mit 0.5 mg Ranibizumab nach Stabilisierung (N=105)
Gruppe II mit 0.5 mg Ranibizumab nach Krankheitsaktivität (N=116)
Gruppe III mit Visudyne-PDT (N=55)
Die in die vPDT-Gruppe randomisierten Patienten konnten ab Monat 3 eine Behandlung mit Ranibizumab erhalten.
Die Visusverbesserung war von einer Reduzierung der zentralen Netzhautdicke begleitet.
In den Ranibizumab-Behandlungsarmen wurde gegenüber der vPDT-Gruppe (p-Wert <0.05) ein von den Patienten subjektiv angegebener Nutzen in Bezug auf die Verbesserung des kombinierten Ergebnisses wie auch des Ergebnisses in mehreren Subskalen (allgemeiner Visus, Nahsichtaktivitäten, psychische Verfassung und unabhängiger Funktionsstatus) des Fragebogens VFQ-25 festgestellt.
Behandlung der RPM bei Frühgeborenen: Studie H2301 (RAINBOW)
Die klinische Sicherheit und Wirksamkeit von Lucentis 0.1 mg bei der Behandlung der RPM bei Frühgeborenen wurde anhand der Sechsmonatsdaten aus der randomisierten, offenen, dreiarmigen Parallelgruppen-Überlegenheitsstudie H2301 (RAINBOW) untersucht, die darauf ausgelegt war, die Anwendung von Ranibizumab in Dosen von 0.1 mg und 0.2 mg, verabreicht als intravitreale Injektionen, im Vergleich zur Lasertherapie zu beurteilen. Geeignete Patienten mussten einen der folgenden Netzhautbefunde in beiden Augen aufweisen:
- Zone I, Krankheitsstadium 1+, 2+, 3 oder 3+
- Zone II, Krankheitsstadium 3+
- Aggressive posteriore RPM (AP-RPM)
Für diese Studie wurden 225 Patienten im Verhältnis 1:1:1 randomisiert, um Ranibizumab intravitreal 0.1 mg (n=77) bzw. 0.2 mg (n=74) oder eine Lasertherapie (n=74) zu erhalten.
Der Behandlungserfolg, gemessen anhand des Ausbleibens einer aktiven ROP und des Fehlens ungünstiger struktureller Auswirkungen in beiden Augen 24 Wochen nach der ersten Studienbehandlung, betrug 75% in der Ranibizumab 0.1 mg-Gruppe und 66.2% in der Lasertherapie-Gruppe. Die Mehrheit der mit Ranibizumab 0.1 mg behandelten Patienten (77.6%) erhielt eine einzige Injektion pro Auge.
Aus der 0.1 mg Ranibizumab-Behandlungsgruppe wechselten weniger Patienten aufgrund fehlenden Ansprechens zu einem anderen Behandlungsmodus als in der Lasertherapie-Gruppe (16.9% vs. 24.3%). Ungünstige strukturelle Auswirkungen waren in der 0.1 mg Ranibizumab-Behandlungsgruppe (5 Patienten, 6.7%) weniger häufig als in der Lasertherapie-Gruppe (7 Patienten, 10.1%). Darüber hinaus erreichten 75% der Patienten innerhalb von 8 Tagen ein Abklingen der Erkrankung unter Ranibizumab 0.1 mg im Vergleich zu 22.5 Tagen in der Lasertherapie-Gruppe.
Pharmakokinetik
Absorption
Die monatliche, intravitreale Verabreichung von Lucentis bei Patienten mit neovaskulärer AMD führt zu allgemein tiefen Serumkonzentrationen von Ranibizumab mit der maximalen Serumkonzentration (Cmax) deutlich unter der Konzentration, welche zu einer 50%igen Hemmung der VEGFs führte (11-27 ng/ml im Zellproliferations-Assay).
Distribution
Die maximale Serumkonzentration (Cmax) liegt im Allgemeinen in einem Bereich von 0.46 bis 1.76 ng/ml, die minimale Serumkonzentration (Cmin) im Bereich von 0.04 bis 0.29 ng/l. Die Cmax im Serum war über einen Dosisbereich von 0.05 bis 1.0 mg/Auge proportional zur verabreichten Dosierung. Die Serumkonzentration in Patienten mit DME und RVO war ähnlich der in Patienten mit neovaskulärer AMD.
Metabolismus
Nicht zutreffend.
Elimination
Basierend auf Ranibizumab Serumkonzentrationen liegt die durchschnittliche Eliminationshalbwertzeit von Ranibizumab im Glaskörper bei Patienten mit neovaskulärer AMD bei etwa 9 Tagen. Es wird angenommen, dass die Serumkonzentrationen von Ranibizumab 90'000-mal tiefer sind als im Glaskörper.
Leberfunktionsstörungen
Es liegen keine spezifischen Untersuchungen vor.
Nierenfunktionsstörungen
Es wurden keine prospektiven Studien zur Pharmakokinetik von Lucentis bei Patienten mit eingeschränkter Nierenfunktion durchgeführt. In einer Populationsanalyse pharmakokinetischer Daten bei Patienten mit neovaskulärer AMD wiesen 68% (N=136/200) der Patienten eine eingeschränkte Nierenfunktion auf (46.5% leicht, 20% mässig und 1.5% schwer). Bei den untersuchten Patienten mit RVO hatten 48.2% (N=253/525) eine eingeschränkte Nierenfunktion (36.4% leicht, 9.5% mittel und 2.3% schwer). Die beobachtete Reduktion der systemischen Clearance von Ranibizumab war statistisch nicht signifikant.
Pädiatrische Population (Frühgeborene mit RPM)
Nach intravitrealer Verabreichung von Ranibizumab 0.1 mg an jedem Auge bei Frühgeborenen mit einer RPM lag die Serumkonzentration von Ranibizumab höher als bei erwachsenen Patienten mit neovaskulärer AMD unter der Behandlung mit 0.5 mg in jedem Auge. Gemäss einer pharmakokinetischen Analyse der Population waren die Unterschiede bei der Cmax und der AUCinf ca. 8- bzw. 5-fach höher. Die scheinbare systemische Halbwertszeit betrug ca. 6 Tage. Bei dieser Analyse wurde kein Zusammenhang zwischen der systemischen Konzentration von Ranibizumab und derjenigen des VEGF festgestellt.
Präklinische Daten
Die bilaterale intravitreale Verabreichung von Ranibizumab an Cynomolgus-Affen in Dosierungen von 0.25 mg/Auge bis zu 2.0 mg/Auge alle 2 Wochen während bis zu 26 Wochen ergab dosisabhängige okuläre Effekte.
In der Vorderkammer wurde ein dosisabhängiges Ansteigen von «Flare» und Zellen beobachtet, mit einem Maximum 2 Tage nach der Injektion. Die Intensität der Entzündungsreaktion verminderte sich üblicherweise bei folgenden Injektionen oder in der Erholungsphase, war aber nicht in allen Fällen vollständig reversibel. In der hinteren Kammer kam es zu reversiblen fokalen perivenösen retinalen Blutungen, die hauptsächlich nach der ersten Verabreichung auftraten, und zu vitrealen Zellinfiltrationen und „mouches volantes“, die tendenziell dosisabhängig waren und bis zum Behandlungsende persistierten. In der Studie über 26 Wochen nahm die Intensität der Entzündung mit der Anzahl Injektionen zu und der Versuch musste frühzeitig abgebrochen werden. Eine Reversibilität der Befunde zeichnete sich in der Erholungsphase ab. Eine Kataraktbildung wurde bei einigen Tieren nach einer relativ langen Phase starker Entzündung beobachtet, was darauf hindeutet, dass die Linsenveränderungen eher sekundär auf die schwere Entzündung folgten. Ein transienter Anstieg des Augeninnendruckes nach Verabreichung wurde unabhängig von der Dosis nach intravitrealer Injektion beobachtet.
Die mikroskopischen Veränderungen in den Augengeweben wurden alle mit der Entzündung in Zusammenhang gebracht und deuteten nicht auf degenerative Prozesse in irgendwelchen Augenstrukturen hin. In einigen Fällen wurden granulomatöse entzündliche Veränderungen am Sehnervenkopf beobachtet. Diese Veränderungen des posterioren Segmentes nahmen während der Erholungsphase ab und verschwanden in einigen Fällen ganz.
Anschliessend an die intravitreale Injektion wurden bei Serumspiegeln, die mehr als 100 fach höher waren als die die bei normaler therapeutischer Anwendung beim Menschen auftreten, keine Zeichen einer systemischen Toxizität beobachtet. Antikörper im Serum, bzw. im Glaskörper gegen Ranibizumab wurden nicht bei allen der behandelten Tieren gefunden.
Die präklinischen Unterlagen decken lediglich die systemische Exposition nach intravitrealer Anwendung ab. Es bestehen keine Daten über die Mutagenität, Karzinogenität und Immunotoxizität von Lucentis bei Tieren.
Reproduktionstoxizität
Bei der intravitrealen Behandlung von trächtigen Affenweibchen mit Ranizumab mit einer maximalen systematischen Exposition, entsprechend dem 0.9-7-Fachen der schlimmstenfalls anzunehmenden klinischen Exposition, wurden keine embryo/fetotoxischen oder teratogenen Effekte gefunden, und es kam zu keiner Auswirkung auf Gewicht und Struktur der Plazenta; trotzdem sollte Ranibizumab aufgrund seines pharmakologischen Effekts als potenziell teratogen und embryo-/fetotoxisch angesehen werden.
Das Fehlen von Ranibizumab/vermittelten Effekten auf die embryo-fötale Entwicklung ist wohl hauptsächlich darauf zurückzuführen, dass das Fab-Fragment die Plazentaschranke nicht passieren kann. Dennoch wurde ein Fall mit hohen maternalen Ranibizumab-Serumspiegeln und der Nachweisbarkeit von Ranibizumab in fötalem Serum beschrieben; dies lässt darauf schliessen, dass der Anti-Ranibizumab-Antikörper als Trägerprotein (mit Fc-Region) für Ranibizumab fungierte, auf diese Weise die Clearance im Serum der Mutter verminderte und das Passieren der Plazentaschranke ermöglichte. Da die Untersuchungen zur embryo-fötalen Entwicklung an gesunden trächtigen Tieren durchgeführt wurden und die Durchgängigkeit der Plazenta für ein Fab-Fragment durch eine Erkrankung modifiziert werden könnte, sollten die Studienergebnisse mit Vorsicht beurteilt werden.
Sonstige Hinweise
Inkompatibilitäten
Nicht zutreffend.
Da keine Verträglichkeitsstudien durchgeführt wurden, darf das Arzneimittel nicht mit anderen Arzneimitteln gemischt werden.
Haltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
Das Arzneimittel soll für Kinder unerreichbar aufbewahrt werden.
Durchstechflasche: In der Originalpackung aufbewahren, um den Inhalt vor Licht zu schützen. Im Kühlschrank (2-8°C) lagern. Nicht einfrieren.
Die ungeöffnete Durchstechflasche kann vor Anwendung bis zu 24 Stunden bei Raumtemperatur (25°C) aufbewahrt werden.
Fertigspritze: In der versiegelten Blisterpackung in der Originalpackung aufbewahren, um den Inhalt vor Licht zu schützen. Im Kühlschrank (2-8°C) lagern. Nicht einfrieren.
Hinweise für die Handhabung
Aus mikrobiologischen Gründen ist die gebrauchsfertige Injektionslösung einer Durchstechflasche oder Fertigspritze unmittelbar nach Anbruch zu verwenden. Die nicht gebrauchte Lösung ist zu verwerfen (s. «Dosierung/Anwendung»). Eine Durchstechflasche oder Fertigspritze ist zur Verabreichung einer Einzeldosis bestimmt.
Durchstechflasche (Erwachsene und Frühgeborene):
Die Durchstechflasche ist steril. Die Durchstechflasche darf nicht verwendet werden wenn die Verpackung beschädigt ist. Die Sterilität der Durchstechflasche kann nur bei intakter Versiegelung der Verpackung garantiert werden. Die Durchstechflasche darf nicht verwendet werden, falls die Lösung verfärbt oder trübe erscheint, oder Partikel enthält.
Zur Vorbereitung und Durchführung der intravitrealen Injektion werden die folgenden Komponenten für die einmalige Benutzung benötigt:
- 5 µm Filternadel (18G)
- Sterile 1 ml Spritze
- Injektionsnadel (13 mm, 30 Gauge)
Diese Komponenten sind nicht in der Packung enthalten, welche nur die Durchstechflasche enthält. Die 1 ml sterile Spritze sowie die Injektionsnadel sind nicht in der Packung enthalten, welche die Durchstechflasche und die Filternadel enthält.
Fertigspritze: Die ungeöffnete Lucentis Fertigspritze kann vor Gebrauch bis zu 24 Stunden bei Raumtemperatur (25°C) aufbewahrt werden.
Fertigspritze (Nur für Erwachsene)
Die Fertigspritze ist nur für die einmalige Anwendung bestimmt (siehe Abschnitt 4 «Dosierung/Anwendung»).
Die Fertigspritze ist steril. Verwenden Sie die Fertigspritze nicht, wenn die Verpackung beschädigt ist. Die Sterilität der Fertigspritze kann nur bei intakter, versiegelter Blisterpackung garantiert werden. Verwenden Sie die Fertigspritze nicht, wenn die Lösung verfärbt oder trüb ist oder Feststoffe enthält.
Zur Vorbereitung von Lucentis für die intravitreale Anwendung folgen Sie bitte der Gebrauchsanweisung:
Überschrift | Anweisungen | Diagramm/Abbildung |
|---|---|---|
Lesen Sie vor Gebrauch der Fertigspritze alle Anweisungen aufmerksam durch. Die Fertigspritze ist nur für die einmalige Anwendung bestimmt. Die Fertigspritze ist steril. Ein Produkt, dessen Verpackung beschädigt ist, darf nicht verwendet werden. Hinweis: Die Dosis muss auf 0.05 ml eingestellt werden. | ||
Beschreibung der Fertigspritze |
| |
Vorbereitung | 1. Vergewissern Sie sich, dass Ihre Verpackung den folgenden Artikel enthält:
2. Ziehen Sie den Deckel der Blisterpackung, in der sich die Spritze befindet, ab und nehmen Sie die Spritze unter aseptischer Handhabung vorsichtig heraus. | (kein Diagramm/keine Abbildung) |
Überprüfen der Spritze | 3. Überprüfen Sie Folgendes:
4. Trifft einer der oben genannten Punkt nicht zu, entsorgen Sie die Fertigspritze und verwenden Sie eine Neue. | |
Abnehmen der Spritzenkappe | 5. Entfernen Sie die Spritzenkappe, indem Sie sie zur Seite hin abknicken (keine Dreh- oder Rotationsbewegung; siehe Abbildung 2). 6. Entsorgen Sie die Spritzenkappe (siehe Abbildung 3). |
|
Befestigen der Kanüle | 7. Bringen Sie eine ca. 1.3 cm lange, sterile Injektionskanüle der Grösse 30G fest an der Spritze an, indem Sie sie bis zum Anschlag auf den Luer-Lock-Anschluss aufschrauben (siehe Abbildung 4). 8. Entfernen Sie vorsichtig die Schutzhülle der Kanüle, indem Sie sie in einer geraden Bewegung abziehen (siehe Abbildung 5). Hinweis: Wischen Sie die Kanüle keinesfalls ab. |
|
Entfernen von Luftblasen | 9. Halten Sie die Spritze senkrecht. 10. Sollten Luftblasen vorhanden sein, klopfen Sie vorsichtig mit dem Finger an die Spritze, bis die Luftblasen nach oben steigen (siehe Abbildung 6). |
|
Einstellen der Dosis | 11. Halten Sie die Spritze auf Augenhöhe und drücken Sie vorsichtig den Kolben, bis sich der Rand unterhalb der Kuppel des Gummistopfens auf einer Linie mit der Dosismarkierung befindet (siehe Abbildung 7).
Hinweis: Der Kolben ist nicht mit dem Gummistopfen verbunden - dadurch wird verhindert, dass Luft in die Spritze gesaugt wird. |
|
Injektion | Die Injektion ist unter aseptischen Bedingungen durchzuführen. 12. Die Injektionskanüle sollte 3.5–4.0 mm hinter dem Limbus in den Glaskörper eingeführt werden. Dabei den waagrechten Meridian meiden und in Richtung des Zentrums des Augapfels zielen. 13. Injizieren Sie die Lösung langsam, bis der Gummistopfen den Boden der Spritze erreicht hat, um das Volumen von 0.05 ml zu verabreichen. 14. Für anschliessende Injektionen sollte eine andere Stelle auf der Lederhaut verwendet werden. 15. Verschliessen Sie nach der Injektion die Kanüle nicht mit der Schutzkappe und entfernen Sie die Kanüle nicht von der Fertigspritze. Entsorgen Sie die benutzte Fertigspritze zusammen mit der Kanüle in einer stichfesten Entsorgungsbox oder nach den lokalen Bestimmungen. | |






Zulassungsnummer
57664, 63277 (Swissmedic)
Zulassungsinhaberin
Novartis Pharma Schweiz AG, Risch; Domizil 6343 Rotkreuz
Stand der Information
Oktober 2020.
Composizione
Principi attivi
Ranibizumab.
Sostanze ausiliarie
α,α‑trealosio diidrato, istidina, istidina cloridrato monoidrato, polisorbato 20, acqua per preparazioni iniettabili.
Forma farmaceutica e quantità di principio attivo per unità
Flaconcino con tappo perforabile: ranibizumab 10 mg/ml (flaconcino contenente 2.3 mg di ranibizumab in 0.23 ml di soluzione).
Siringa preriempita: ranibizumab 10 mg/ml (siringa preriempita contenente 1.65 mg di ranibizumab in 0.165 ml di soluzione).
Indicazioni/Possibilità d'impiego
Lucentis è indicato negli adulti per:
- il trattamento della degenerazione maculare senile essudativa (AMD essudativa);
- il trattamento della perdita dell'acuità visiva causata da edema maculare diabetico (DME);
- il trattamento della retinopatia diabetica non proliferativa (NPDR) da moderata a grave o della retinopatia diabetica proliferativa (PDR);
- il trattamento della perdita dell'acuità visiva causata da edema maculare secondario a occlusione venosa retinica (di branca, BRVO, o centrale, CRVO);
- il trattamento della diminuzione visiva causata da neovascolarizzazione coroideale (CNV) attiva;
- il trattamento della perdita dell'acuità visiva causata da neovascolarizzazione coroidale (CNV) secondaria a miopia patologica (PM).
Lucentis è indicato nei neonati prematuri per il trattamento della retinopatia del prematuro (ROP):
- ROP nella zona I (stadio della malattia 1+, 2+, 3 o 3+), nella zona II (stadio della malattia 3+) o ROP‑AP (ROP aggressivo posteriore).
Posologia/Impiego
Lucentis deve essere somministrato da un oculista qualificato che disponga di una struttura adeguata. Lucentis si somministra nella camera vitrea (per via intravitreale). Un flaconcino con tappo perforabile di Lucentis (adulti e neonati prematuri) contiene 0.23 ml; una siringa preriempita (solo per adulti), 0.165 ml: entrambi sono destinati a essere utilizzati una sola volta per un solo paziente.
Posologia abituale
Posologia negli adulti
La dose raccomandata di Lucentis è 0.5 mg somministrata come singola iniezione intravitreale. Ciò corrisponde a un volume iniettato di 0.05 ml. L'intervallo tra due dosi iniettate nello stesso occhio deve essere di almeno un mese.
Il trattamento viene iniziato con un'iniezione al mese fino ad ottenere la massima acuità visiva e/o non vi sono più segni di attività della malattia. Nei pazienti affetti da AMD essudativa, DME, NPDR da moderata a grave o PDR, BRVO o CRVO, può essere necessario iniziare la terapia con tre o più iniezioni mensili consecutive.
Pertanto, gli intervalli di monitoraggio e di trattamento devono essere stabiliti dal medico. In occasione delle visite di controllo, l'attività della malattia viene valutata per mezzo di esame clinico, valutazione dell'acuità visiva e/o esami di imaging (ad esempio tomografia a coerenza ottica e/o angiografia con fluoresceina).
Gli intervalli di monitoraggio e trattamento possono essere estesi o ridotti gradualmente in funzione dell'attività della malattia e del decorso terapeutico osservato.
Se il medico lo ritiene indicato, Lucentis può essere somministrato anche mensilmente su un dato occhio.
Il trattamento della perdita dell'acuità visiva causata da CNV deve essere stabilito caso per caso sulla base dell'attività della malattia. Per la CNV secondaria a miopia patologica (PM) o per altre CNV che non insorgono nel contesto di una AMD, non vi sono esperienze che vadano oltre l'intervallo di trattamento di un anno.
Posologia nei neonati prematuri
La posologia raccomandata di Lucentis nei neonati prematuri è di 0.1 mg somministrata come singola iniezione intravitreale. Ciò corrisponde a un volume di iniezione di 0.01 ml. Nei neonati prematuri, il trattamento della retinopatia del prematuro (ROP) viene iniziato con una singola dose che può essere somministrata in entrambi gli occhi nello stesso giorno. In totale, possono essere somministrate fino a tre iniezioni per occhio in sei mesi se ci sono segni di attività della malattia. Se è necessaria più di un'iniezione, l'intervallo tra due iniezioni deve essere di almeno un mese.
Pazienti con disturbi della funzionalità epatica
Non sono disponibili studi su pazienti con insufficienza epatica. Tuttavia, essendo la quantità disponibile per via sistemica trascurabile, non sono necessarie speciali misure precauzionali per questa popolazione.
Pazienti con disturbi della funzionalità renale
Non è necessario un aggiustamento della dose nei pazienti con insufficienza renale (cfr. il capitolo «Farmacocinetica»).
Pazienti anziani (da 65 anni in su)
Non si richiedono aggiustamenti della dose.
Bambini e adolescenti
Non sono disponibili dati sufficienti sulla sicurezza e l'efficacia di Lucentis nei bambini e negli adolescenti e, pertanto, se ne sconsiglia l'impiego in questa popolazione di pazienti. I dati disponibili in pazienti adolescenti di età compresa tra 12 e 17 anni con perdita dell'acuità visiva secondaria a CNV sono limitati (cfr. il capitolo «Avvertenze e misure precauzionali»).
Modo di somministrazione
Prima di somministrare la terapia intravitreale, si devono acquisire dati anamnestici completi con riferimento a possibili reazioni di ipersensibilità (cfr. il capitolo «Avvertenze e misure precauzionali»).
La somministrazione di Lucentis deve essere effettuata in condizioni asettiche (locale idoneo, telino sterile, guanti sterili, strumenti sterili). Prima dell'iniezione devono essere somministrati un'anestesia adeguata e un antimicrobico topico ad ampio spettro per disinfettare la cute perioculare, la palpebra e la superficie oculare.
L'intero contenuto del flaconcino viene prelevato con una siringa e un ago filtro da 5 µm. Prima dell'iniezione intravitreale, rimuovere l'ago filtro e applicare sulla siringa l'ago per iniezione (13 mm, 30 gauge). Aspirare il contenuto del flaconcino fino a che la testa dello stantuffo della siringa è allineata con la tacca indicante 0.05 ml (50 µl). Il contenuto di un flaconcino è destinato alla somministrazione di una sola dose. Smaltire la soluzione inutilizzata.
Negli adulti, l'ago per iniezione deve essere inserito completamente 3.5–4.0 mm posteriormente al limbus nella camera vitreale, evitando il meridiano orizzontale e dirigendo l'ago verso il centro del globo oculare.
Iniettare lentamente il volume d'iniezione; per le iniezioni successive scegliere un altro punto della sclera.
Dopo l'iniezione, tenere sotto controllo la pressione intraoculare del paziente. Il monitoraggio del paziente deve comprendere il controllo della perfusione della testa del nervo ottico subito dopo l'iniezione, la misurazione della pressione intraoculare dopo 30 minuti, esame oftalmoscopico, esame con la lampada a fessura ed esame del fondo dell'occhio dopo 2–7 giorni. Al paziente deve essere raccomandato di segnalare immediatamente eventuali segni di endoftalmite (cfr. il capitolo «Avvertenze e misure precauzionali»).
Nei neonati prematuri, l'ago per iniezione deve essere inserito 1.0–2.0 mm posteriormente al limbus, dirigendo l'ago verso il nervo ottico. Quindi, viene somministrato il volume di iniezione pari a 0.01 ml.
Controindicazioni
Ipersensibilità al principio attivo o a una qualsiasi delle sostanze ausiliarie. Lucentis è controindicato nei pazienti con infezioni oculari o perioculari, come pure nei pazienti con infiammazioni intraoculari in atto.
Avvertenze e misure precauzionali
Reazioni correlate all'iniezione intravitreale
Le iniezioni intravitreali possono essere associate a endoftalmite infettiva e a distacco di retina. Per la somministrazione di Lucentis devono sempre essere usate idonee tecniche di iniezione in asepsi. Inoltre, i pazienti devono essere tenuti sotto osservazione nei giorni successivi all'iniezione in modo da avviare subito un trattamento in caso di infezione (cfr. il capitolo «Effetti indesiderati»).
Negli adulti, entro 60 minuti dall'iniezione di Lucentis, sono stati osservati aumenti transitori della pressione intraoculare, che in alcuni casi sono stati anche persistenti. La pressione intraoculare e la perfusione dell'arteria centrale della retina devono essere controllate e trattate in modo appropriato (cfr. il capitolo «Effetti indesiderati»).
Trattamento bilaterale
L'efficacia e la sicurezza del trattamento bilaterale concomitante con Lucentis non sono state valutate in studi appositi.
I dati limitati sull'uso bilaterale di Lucentis (incl. la somministrazione nello stesso giorno) non evidenziano un aumento del rischio di eventi avversi sistemici rispetto al trattamento unilaterale.
Eventi tromboembolici arteriosi
Esiste un rischio potenziale di eventi tromboembolici arteriosi in seguito a iniezione intravitreale di inibitori del VEGF (dall'inglese vascular endothelial growth factor, fattore di crescita dell'endotelio vascolare). Il rischio è presumibilmente elevato nei pazienti con rischio noto per ictus (ad es. pazienti con ictus o attacco ischemico transitorio nell'anamnesi).
Immunogenicità
In tutti i gruppi sottoposti al trattamento, lo 0–3% dei pazienti non ancora trattati ha evidenziato una reazione immunitaria verso Lucentis. Quando Lucentis è stato somministrato mensilmente, dopo 12–24 mesi il titolo anticorpale è risultato basso nell'1–6% dei pazienti. Questi dati relativi all'immunogenicità indicano la percentuale di pazienti risultati positivi al test con elettrochemiluminescenza. I dati hanno risentito notevolmente della sensibilità e della specificità del test. La significatività clinica della risposta immunitaria a Lucentis non è ancora chiara. Alcuni pazienti che avevano evidenziato i titoli più elevati di immunoreattività hanno evidenziato irite e disturbi del vitreo.
Popolazioni di pazienti con dati limitati
L'impiego di Lucentis non è stato ancora studiato in pazienti con infezioni sistemiche attive o con altre patologie oculari quali distacco di retina o foro maculare.
Lucentis e fotocoagulazione laser
È stata studiata sia la somministrazione di Lucentis in pazienti precedentemente sottoposti alla fotocoagulazione laser, sia la somministrazione di Lucentis in concomitanza con la fotocoagulazione laser. Quando somministrato nello stesso giorno, Lucentis deve essere somministrato almeno 30 minuti dopo la fotocoagulazione laser.
Durata del trattamento
Per la CNV secondaria a miopia patologica (PM) o per altre CNV che non insorgono nel contesto di una AMD, non vi sono esperienze che vadano oltre l'intervallo di trattamento di un anno.
Interazioni
Non sono stati condotti studi particolari d'interazione.
Gravidanza/Allattamento
Gravidanza
Non sono disponibili dati sull'utilizzo di ranibizumab in gravidanza.
Gli studi su scimmie cynomolgus non hanno mostrato una tossicità diretta o indiretta sulla gravidanza o sullo sviluppo embrionale/fetale (cfr. il capitolo «Dati preclinici»). Ranibizumab inibisce il fattore VEGF‑A, un elemento importante per l'angiogenesi nella fase di sviluppo embriofetale e di formazione della placenta. L'esposizione sistemica al ranibizumab dopo somministrazione oculare è modesta, ma a causa del meccanismo d'azione deve essere considerato come potenzialmente teratogeno ed embriofetotossico. Pertanto, alle donne che pianificano una gravidanza si raccomanda di aspettare 3 mesi prima di concepire un bambino.
Donne in età fertile
Le donne in età fertile devono usare metodi contraccettivi efficaci durante il trattamento e fino a 30 giorni dopo la conclusione.
Allattamento
Non è noto se Lucentis venga escreto nel latte materno. Poiché sono numerose le sostanze che passano nel latte materno e siccome sussiste la possibilità che il medicamento venga assorbito con conseguente compromissione della crescita e dello sviluppo del bambino, si raccomanda di non allattare durante la terapia con Lucentis.
Fertilità
Non sono disponibili dati sull'influsso di Lucentis sulla fertilità maschile e femminile.
Effetti sulla capacità di condurre veicoli e sull'impiego di macchine
Il trattamento con Lucentis può indurre disturbi visivi transitori che possono compromettere la capacità di guidare o la capacità di utilizzare macchine (cfr. il capitolo «Effetti indesiderati»). I pazienti che manifestano questi sintomi non devono guidare o utilizzare macchine fino a quando questi disturbi visivi transitori non scompaiono.
Effetti indesiderati
La base per le considerazioni sulla sicurezza di Lucentis deriva da una popolazione complessiva di 1315 pazienti che hanno partecipato a tre studi clinici di fase III sul trattamento della AMD essudativa. Tutti i pazienti hanno ricevuto Lucentis per almeno 24 mesi; la dose raccomandata di 0.5 è stata somministrata a 440 pazienti.
Gli effetti indesiderati gravi correlati alla procedura di iniezione sono stati: endoftalmite, distacco di retina regmatogeno, rottura retinica e cataratta traumatica iatrogena (cfr. il capitolo «Avvertenze e misure precauzionali»).
Nei pazienti trattati con Lucentis sono stati osservati anche infiammazione intraoculare e aumento della pressione intraoculare (cfr. il capitolo «Avvertenze e misure precauzionali»).
Nei pazienti con AMD essudativa trattati con 0.5 mg di Lucentis nell'ambito degli studi controllati di fase III per AMD essudativa FVF2598g (MARINA), FVF2587g (ANCHOR) e FVF3192g (PIER), gli effetti indesiderati riportati più avanti sono risultati più frequenti (almeno 2 punti percentuali) rispetto a quanto osservato nei gruppi di controllo [iniezione sham (cfr. il capitolo «Proprietà/effetti») oppure di terapia fotodinamica (PDT) con verteporfina]. Di conseguenza sono stati descritti come potenziali effetti indesiderati. I dati sulla sicurezza citati sotto considerano, tra l'altro, tutti gli effetti indesiderati che si suppone siano riconducibili almeno potenzialmente all'iniezione in sé o al medicamento e che si sono manifestati nei 440 pazienti che hanno ricevuto 0.5 mg di Lucentis nell'ambito di una terapia di combinazione per il trattamento dell'AMD essudativa.
Popolazione di pazienti con DR
La sicurezza di Lucentis è stata valutata in uno studio clinico di 24 mesi nello studio Protocol‑S, che includeva, tra gli altri, 191 pazienti trattati con ranibizumab con retinopatia diabetica (DR). Gli eventi oculari e non oculari osservati erano coerenti con quanto ci si aspetterebbe in una popolazione di pazienti diabetici con DR, o erano simili in termini di frequenza e gravità agli eventi osservati in precedenti studi clinici condotti su Lucentis.
Popolazione di pazienti con CNV
La sicurezza di Lucentis è stata valutata nell'ambito di uno studio clinico di 12 mesi (MINERVA) cui hanno preso parte 171 pazienti trattati con ranibizumab che avevano perso la vista a seguito di CNV (cfr. il capitolo «Proprietà/effetti»). Il profilo di sicurezza osservato in questi pazienti è risultato coerente con quello di precedenti studi clinici su Lucentis.
Popolazione di pazienti con CNV secondaria a PM
La sicurezza di Lucentis è stata valutata nell'ambito dello studio clinico RADIANCE di 12 mesi. A questo studio hanno preso parte 224 pazienti con CNV secondaria a PM, i quali sono stati trattati con ranibizumab (cfr. il capitolo «Proprietà/effetti»). Frequenza e gravità delle reazioni avverse oculari e non oculari verificatesi nell'ambito di questo studio sono state comparabili a quelle registrate negli studi sull'AMD essudativa.
Popolazione con retinopatia del prematuro (ROP)
- La sicurezza di Lucentis 0.1 mg è stata studiata in uno studio clinico di sei mesi (RAINBOW) che ha coinvolto 77 neonati prematuri affetti da ROP trattati con ranibizumab (cfr. il capitolo «Proprietà/effetti»). Gli effetti collaterali a livello oculare osservati nello studio RAINBOW erano coerenti con quelli osservati anche negli adulti trattati con ranibizumab 0.5 mg o che rappresentavano un evento nella ROP. Gli effetti collaterali non oculari in questo studio clinico sono stati generalmente coerenti con quelli attesi in questa popolazione di pazienti con comorbidità multiple dovute al parto pretermine. Teoricamente, si deve considerare che la maturazione di altri organi può essere ritardata dopo il trattamento con anti‑VEGF e quindi possono verificarsi complicazioni.
Classi di frequenza: «Molto comune» (≥1/10), «comune» (≥1/100 <1/10), «non comune» (≥1/1000 <1/100), «raro» (≥1/10'000 <1/1000), «molto raro» (<1/10'000).
Infezioni ed infestazioni
Molto comune: nasofaringite (12.5–16.4%).
Comune: influenza, infezione delle vie urinarie.
Patologie del sistema emolinfopoietico
Comune: anemia.
Disturbi del sistema immunitario
Comune: reazioni di ipersensibilità.
Disturbi psichiatrici
Comune: ansia.
Patologie del sistema nervoso
Molto comune: cefalea (8–15%).
Comune: ictus.
Patologie dell'occhio
Molto comune: infiammazioni intraoculari (10–18%), vitreite (2.3–10%), distacco del vitreo (18–19%), emorragia retinica (25%), disturbi della vista (6.6–10.5%), dolore oculare (27–32%), mosche volanti (7–25%), emorragia congiuntivale (55–72%), irritazione oculare (12–15%), sensazione di corpo estraneo nell'occhio (12–15%), lacrimazione aumentata (10–14%), blefarite (8–11%), prurito (8.5–10%).
Comune: degenerazione retinica, disturbi della retina, distacco della retina, lacerazione della retina, distacco dell'epitelio pigmentato della retina, lacerazioni dell'epitelio pigmentato della retina, riduzione dell'acuità visiva, emorragie e disturbi del vitreo, uveite, irite, iridociclite, cataratta (subcapsulare), opacizzazione della capsula posteriore, cheratite puntata, abrasioni corneali, opacizzazioni dell'umor acqueo, visione offuscata, emorragie nella sede di iniezione, emorragie oculari, congiuntivite (allergica), secrezioni oculari, fotopsia, fotofobia, fastidio oculare, dolore ed edema palpebrale, iperemia congiuntivale.
Non comune: endoftalmite, ipopion, ifema, cheratopatia, sinechie dell'iride, depositi sulla superficie della cornea, edema corneale, strie di Vogt, dolore e irritazioni nella sede di iniezione, cecità, irritazioni della palpebra.
Patologie cardiache, patologie vascolari
Eventi tromboembolici arteriosi secondo la definizione data dall'Antiplatelet Trialists' Collaboration (1994), comprendenti morte per disturbi vascolari, infarto miocardico non fatale, ictus ischemico non fatale e ictus emorragico non fatale, sono stati messi in correlazione con la disponibilità sistemica di inibitori del VEGF molto potenti. Nel primo anno la percentuale di eventi tromboembolici registrata in ambedue i gruppi di pazienti trattati con Lucentis (0.3 e 0.5 mg) è stata del 2.3%, mentre nel gruppo di controllo di appena l'1.3%. Nel secondo anno dello studio MARINA è stata in ambedue i gruppi del 3.0%, mentre nel gruppo di controllo del 3.2%.
Patologie respiratorie, toraciche e mediastiniche
Comune: tosse.
Patologie gastrointestinali
Comune: nausea.
Patologie della cute e del tessuto sottocutaneo
Non comune: reazioni allergiche (eruzione cutanea, orticaria, prurito, eritema).
Patologie del sistema muscoloscheletrico e del tessuto connettivo
Molto comune: dolore articolare (8–12%).
Esami diagnostici
Pressione intraoculare aumentata.
Immunogenicità
Come per tutte le proteine terapeutiche, sussiste la possibilità di una risposta immunitaria nei pazienti che hanno ricevuto Lucentis. I dati relativi all'immunogenicità riflettono la percentuale di pazienti risultati positivi ai saggi immunologici per la presenza di anticorpi anti-ranibizumab. I dati hanno risentito notevolmente della sensibilità e della specificità dei saggi.
Negli studi sull'AMD, in tutti i gruppi sottoposti al trattamento lo 0–3% dei pazienti non ancora trattati ha evidenziato una reazione immunitaria verso Lucentis. Quando Lucentis è stato somministrato mensilmente per più di 12–24 mesi, la presenza di anticorpi verso ranibizumab è stata rilevata approssimativamente nell'1–8% dei pazienti affetti da AMD neovascolare.
Negli studi sul DME, in tutti i gruppi sottoposti al trattamento lo 0–2% dei pazienti non ancora trattati ha evidenziato una reazione immunitaria verso Lucentis. Quando Lucentis è stato somministrato mensilmente per più di 12 mesi, la presenza di anticorpi verso ranibizumab è stata rilevata approssimativamente nel 2–4% dei pazienti affetti da DME.
Negli studi sull'RVO, in tutti i gruppi sottoposti al trattamento il 2–3% dei pazienti non ancora trattati ha evidenziato una reazione immunitaria verso Lucentis. Quando Lucentis è stato somministrato mensilmente per più di 12 mesi, la presenza di anticorpi verso ranibizumab è stata rilevata approssimativamente nel 4–5% dei pazienti affetti da RVO.
La significatività clinica dell'immunoreattività verso Lucentis non è ancora chiara.
Secondo i dati aggregati ottenuti dagli studi clinici randomizzati, in doppio cieco, portati a termine, nei pazienti con DME le infezioni/infiammazioni non gravi della ferita in sede non oculare osservate in terapia con ranibizumab 0.5 mg sono risultate più frequenti che nel gruppo di controllo (1.85/100 anni paziente vs. 0.27/100 anni paziente). Non è ancora nota la correlazione con ranibizumab. Sussiste, tuttavia, il rischio teorico di una correlazione tra questi eventi e l'impiego di inibitori del VEGF.
La notifica di effetti collaterali sospetti dopo l'omologazione del medicamento è molto importante. Consente una sorveglianza continua del rapporto rischio-benefico del medicamento. Chi esercita una professione sanitaria è invitato a segnalare qualsiasi nuovo o grave effetto collaterale sospetto attraverso il portale online ElViS (Electronic Vigilance System). Maggiori informazioni sul sito www.swissmedic.ch
Posologia eccessiva
Gli studi clinici sull'AMD essudativa e i dati post-marketing hanno riportato casi di sovradosaggio accidentale (iniezione di volumi più alti di quello raccomandato di 0.05 ml di Lucentis).
Segni e sintomi
Le reazioni avverse più frequenti sono state innalzamento della pressione intraoculare e dolore oculare.
Trattamento
Se si verifica un sovradosaggio, si deve controllare la pressione intraoculare e, a discrezione del medico curante, avviare eventualmente il trattamento.
Negli studi clinici, i pazienti con AMD essudativa e DME hanno ricevuto una dose massima di 2 mg di ranibizumab in un volume d'iniezione da 0.05 ml a 0.10 ml. Tipo e frequenza delle reazioni avverse oculari e sistemiche sono stati comparabili a quelli osservati per Lucentis 0.5 mg (in 0.05 ml).
Proprietà/Effetti
Codice ATC
S01LA04
Meccanismo d'azione
Il principio attivo di Lucentis (ranibizumab) è un frammento di un anticorpo monoclonale ricombinante umanizzato (Fab) diretto contro il fattore di crescita endoteliale vascolare umano A (VEGF‑A). Esso si lega con un'elevata affinità al VEGF‑A e alle sue isoforme. Le isoforme, quali ad esempio, VEGF121 e VEGF165, hanno origine da splicing alternativo dell'mRNA, l'isoforma VEGF110 da proteolisi. Il legame tra ranibizumab e il VEGF‑A e le sue isoforme inibisce l'attivazione dei recettori VEGFR‑1 e VEGFR‑2 sulla superficie delle cellule endoteliali.
Farmacodinamica
Dall'attivazione dei recettori VEGFR‑1 e ‑2 derivano proliferazione delle cellule endoteliali, neovascolarizzazione e aumento della permeabilità vascolare. Si ritiene che tutti questi fattori contribuiscano alla progressione della forma neovascolare della degenerazione maculare senile (AMD), allo sviluppo della CNV, inclusa la CNV secondaria a miopia patologica (PM), a edema maculare diabetico (DME) o a occlusione venosa retinica, che possono determinare la perdita dell'acuità visiva (RVO).
Efficacia clinica
Trattamento dell'AMD essudativa
La sicurezza e l'efficacia clinica di ranibizumab per il trattamento dell'AMD essudativa sono state valutate nel contesto di tre studi randomizzati, in doppio cieco, su un totale di 1323 pazienti (Lucentis: n = 879, gruppi di controllo n = 444) con AMD neovascolare. Nello studio MARINA il controllo è stato confrontato versus sham, mentre nello studio ANCHOR il controllo è stato attivo mediante PDT con Visudyne. Sono stati arruolati pazienti con lesioni della dimensione massima di 12 superfici papillari e acuità visiva compresa tra 20/40 e 20/320 (Snellen) nell'occhio oggetto di studio. L'età media dei pazienti era di 77 anni. Negli studi clinici i pazienti hanno ricevuto istruzioni per l'autosomministrazione di un collirio antibiotico (quattro volte al giorno, 3 giorni prima e 3 giorni dopo ogni iniezione).
Nello studio MARINA di 24 mesi sono stati arruolati 716 pazienti con CNV minimamente classica o occulta, i quali sono stati sottoposti al trattamento con iniezione intravitreale di Lucentis 0.3 mg (n = 238), Lucentis 0.5 mg (n = 240) o sham (n = 238). Nei 24 mesi della fase di trattamento i pazienti hanno ricevuto in media 22–24 possibili trattamenti. I risultati ottenuti dallo studio MARINA dopo 12 mesi di trattamento sono stati confermati sostanzialmente nel 90% dei pazienti anche dopo 24 mesi di trattamento (1 volta al mese).
Nello studio ANCHOR di 24 mesi sono stati arruolati 423 pazienti con lesioni da CNV prevalentemente classiche, i quali hanno ricevuto iniezioni intravitreali mensili di Lucentis 0.3 mg e PDT sham (n = 143), iniezioni intravitreali di Lucentis 0.5 mg e PDT sham (n = 140) oppure iniezioni intravitreali sham e PDT con verteporfina attiva (n = 143). La prima PDT sham e con verteporfina attiva sono state applicate contemporaneamente all'iniezione iniziale di Lucentis. Successivamente il trattamento è stato effettuato ogni 3 mesi se la fluorangiografia evidenziava una persistenza o ripresa della permeabilità vascolare nell'occhio oggetto di studio. I risultati sono riassunti nelle tabelle seguenti:
Tabella 0-1 - Studio MARINA: risultati a 12 e 24 mesi
Variazione dell'acuità visiva (in lettere, ETDRS) | Mese | Sham | Ranibizumab 0.5 mg(n = 240) | Differenza | ||
Perdita di < 15 lettere nell'acuità visiva (%)b | Mese 12 | 62% | 95% | 32% | ||
Mese 24 | 53% | 90% | 37% | |||
Aumento ≥15 lettere nell'acuità visiva (%)b | Mese 12 | 5% | 34% | 29% | ||
Mese 24 | 4% | 33% | 29% | |||
Variazione media dell'acuità visivab (lettere) | Mese 12 | -10.5 (16.6) | +7.2 (14.4) | 17.5 | ||
Mese 24 | -14.9 (18.7) | +6.6 (16.5) | 21.1 | |||
a dopo stratificazione b p < 0.01 | ||||||
Tabella 0-2 - Studio ANCHOR: risultati a 12 e 24 mesi
Variazione dell'acuità visiva | PDT con verteporfina | Ranibizumab 0.5 mg | Differenza | |
|---|---|---|---|---|
| Perdita di <15 lettere nell'acuità visiva (%)b | Mese12 | 64% | 96% | 33% (25%, 41%) |
| Mese24 | 66% | 90% | 25% (16%, 34%) | |
| Aumento ≥15 lettere nell'acuità visiva (%)b | Mese 12 | 6% | 40% | 35% (26%, 44%) |
| Mese 24 | 6% | 41% | 35% (26%, 44%) | |
| Variazione media dell'acuità visivab (lettere) | Mese 12 | -9.5 (16.4) | +11.3 (14.6) | 21.1 (17.5, 24.6) |
| Mese 24 | -9.8 (16.4) | +10.7 (16.5) | 20.7 (16.8, 24.7) | |
a dopo stratificazione b p <0.01 | ||||
Sia nello studio MARINA che nello studio ANCHOR il miglioramento della funzione visiva ha rivelato un beneficio nei pazienti trattati con Lucentis 0.5 mg, come attestato dal punteggio alle tre sottoscale del questionario sulla funzione visiva del National Eye Institute (NEI VFQ‑25) che prima erano state poste come endpoint secondari di efficacia (attività con riferimento alla vista da vicino e da lontano, attività dipendenti dalla capacità visiva). Tutte le differenze tra Lucentis 0.5 mg e i due gruppi di controllo sono risultate statisticamente significative e clinicamente rilevanti con valori di p compresi tra 0.009 e < 0.0001.
Nello studio PIER sono stati arruolati 184 pazienti con lesioni da CNV (con e senza componente classica), i quali hanno ricevuto Lucentis 0.3 mg, Lucentis 0.5 mg o iniezione sham una volta al mese per i primi 3 mesi. Successivamente Lucentis è stato somministrato una volta ogni 3 mesi. Dal mese 14 dello studio, i pazienti trattati con iniezione sham sono passati al trattamento con Lucentis e dal mese 19 le iniezioni di Lucentis sono state effettuate a intervalli più ravvicinati. I pazienti trattati con Lucentis nello studio PIER hanno ricevuto mediamente 10 trattamenti nell'arco di 24 mesi.
L'endpoint primario di efficacia era un miglioramento medio dell'acuità visiva nel corso dei 12 mesi. Dopo un iniziale aumento nell'acuità visiva nel periodo in cui la somministrazione era mensile, l'acuità visiva dei pazienti è peggiorata quando la terapia è diventata trimestrale, ritornando ai valori iniziali dopo 12 mesi. Quest'effetto è stato mantenuto nella maggior parte dei pazienti trattati con Lucentis (82%) al mese 24. I dati ottenuti da un numero limitato di pazienti che dopo un anno di trattamento sham sono passati a Lucentis indicano che l'inizio precoce del trattamento è associato a un migliore mantenimento dell'acuità visiva.
Tabella 0-3 - Studio PIER: risultati a 12 mesi
Variazione dell'acuità visiva | Sham | Ranibizumab 0.5 mg | Differenza |
Perdita di < 15 lettere nell'acuità visiva (%)b | 49 | 90 | 37 (23, 52) |
Aumento ≥15 lettere nell'acuità visiva (%)b | 10 | 13 | 2 (-8, 12) |
Variazione media dell'acuità visivab | -16.3 (22.3) | -0.2 (13.1) | 14.7 (8.2, 21.2) |
a dopo stratificazione b p < 0.0001 | |||
Lo studio di fase IIIb SAILOR è stato condotto su pazienti con CNV secondaria a AMD naïve al trattamento e già trattati. Lo studio SAILOR era multicentrico con durata di 1 anno. L'obiettivo primario è stato la valutazione dell'incidenza delle reazioni avverse oculari e non oculari durante i 12 mesi di trattamento. 2378 pazienti sono stati randomizzati in 2 gruppi a ricevere rispettivamente
Lucentis 0.3 mg e 0.5 mg una volta al mese per 3 mesi; il trattamento è quindi proseguito a intervalli di almeno 1 mese in funzione del riscontro.
In generale non sono state osservate differenze tra i due gruppi per quanto concerne l'insorgenza di reazioni avverse oculari e non oculari. In particolare, non sono state osservate differenze tra i due gruppi con riferimento al numero di ictus. Nel gruppo trattato con 0.3 mg, gli ictus sono stati 8/1169 pazienti (0.7%, IC 95%: 0.3–1.3%); in quello trattato con 0.5 mg, 15/1209 pazienti (1.2%, IC 95%: 0.7–2%). Si presume che i pazienti con fattori di rischio noti - tra cui, ad esempio, ictus o TIA nell'anamnesi - abbiano un alto rischio di ictus durante il trattamento con Lucentis.
Trattamento della perdita dell'acuità visiva secondaria a DME
La sicurezza e l'efficacia clinica di Lucentis in pazienti con perdita dell'acuità visiva secondaria a edema maculare diabetico (DME) sono state valutate nell'ambito dello studio RESTORE su un totale di 345 pazienti distribuiti in tre bracci: Braccio 1, 116 pazienti sottoposti inizialmente a iniezione intravitreale di ranibizumab 0.5 mg come monoterapia e fotocoagulazione laser sham; Braccio 2, 118 pazienti sottoposti inizialmente a iniezione intravitreale di ranibizumab 0.5 mg come monoterapia e fotocoagulazione laser; Braccio 3, 111 pazienti sottoposti inizialmente a monoterapia con fotocoagulazione laser e iniezione sham.
Il trattamento con ranibizumab è stato proseguito con iniezioni intravitreali mensili e interrotto qualora la visione del paziente in trattamento con Lucentis avesse evidenziato una stabilizzazione in tre visite di controllo consecutive. Dopo l'interruzione il trattamento è stato ripreso se la visione del paziente fosse peggiorata per via della progressione del DME. In tal caso, la fotocoagulazione laser è stata eseguita nello stesso giorno, almeno 30 minuti dopo la somministrazione di ranibizumab, in linea con i criteri dello studio ETDRS.
I risultati sono riassunti nelle tabelle seguenti:
Tabella 0-4 - Studio RESTORE: risultati a 12 mesi
Variazione della migliore acuità visiva corretta | Ranibizumab | Ranibizumab | Laser | ||
Variazione media in lettere della migliore acuità visiva corretta dal 1º al 12º mese rispetto alla visione all'inizio dello studio (deviazione standard)a | 6.1 (6.4) | 5.9 (7.9) | 0.8 (8.6) | ||
Variazione media in lettere della migliore acuità visiva corretta al 12º mese rispetto alla visione all'inizio dello studio (deviazione standard) | 6.8 (8.3)a | 6.4 (11.8)b | 0.9 (11.4) | ||
Aumento di ≥10 lettere della migliore capacità visita corretta (% di pazienti) | 37.4c | 43.2 | 15.5 | ||
Aumento di ≥15 lettere della migliore capacità visita corretta (% di pazienti) | 22.6d | 22.9e | 8.2 | ||
a p < 0.0001, b p = 0.0004, c p = 0.0001, d p = 0.0032, e p = 0.0021 | |||||
Lo studio di estensione è uno studio in aperto, multicentrico, di 24 mesi. 240 pazienti che avevano completato i 12 mesi dello studio principale sono stati arruolati per l'estensione, nella quale sono stati trattati al bisogno (pro re nata, PRN) con ranibizumab 0.5 mg nello stesso occhio selezionato come oggetto di studio nell'ambito della sperimentazione principale. A seguito di una riduzione nella BCVA secondaria a DME, la somministrazione mensile è stata ripresa e continuata fino a ripristinare un valore stabile della BCVA. Inoltre, se ritenuto necessario dal medico sperimentatore, è stato eseguito il trattamento con il laser in conformità alle linee guida dello studio ETDRS.
Nella fase di estensione di 24 mesi, i pazienti che nello studio principale avevano fatto parte del gruppo di trattamento con ranibizumab hanno ricevuto in media 6.4 iniezioni di ranibizumab. Sui 74 pazienti che nell'ambito dello studio principale avevano costituito il braccio sottoposto a trattamento con il laser, 59 (79%) hanno ricevuto ranibizumab in un qualunque momento dello studio di estensione. Per questi 59 pazienti, il numero medio di iniezioni di ranibizumab ricevute nel corso dei 24 mesi della fase di estensione è stato pari a 8.1. Le percentuali di pazienti che nella fase di estensione non hanno avuto necessità di essere sottoposti al trattamento con ranibizumab sono state del 19%, 25% e 20%, rispettivamente, nei gruppi già trattati con ranibizumab, ranibizumab in combinazione con la laserterapia e laser soltanto. I risultati sono riassunti nella tabella seguente:
Tabella 0-5 - Risultati a 36 mesi dello studio di estensione RESTORE
Outcome vs. basale dello studio principale | Pregressa terapia con ranibizumab | Pregressa terapia con ranibizumab | Pregressa laserterapia |
Variazione media della BCVA vs. basale nello studio principale dopo 36 mesi (DS) | 8.0 (10.09) | 6.7 ( 9.59) | 6.0 (9.35) |
Aumento di ≥10 lettere vs. basale nello studio principale e della BCVA ≥84 (%) dopo 36 mesi | 39 (47.0) | 37 (44.6) | 31 (41.9) |
Aumento di ≥15 lettere vs. basale nello studio principale e della BCVA ≥84 (%) dopo 36 mesi | 23 (27.7) | 25 (30.1) | 16 (21.6) |
n = numero di pazienti per i quali sono stati disponibili sia i valori basali (studio principale, mese 0) sia i valori dopo 36 mesi. * 59 (79%) dei 74 pazienti precedentemente sottoposti a trattamento con il laser hanno ricevuto ranibizumab nello studio di estensione. | |||
Il profilo di sicurezza a lungo termine di ranibizumab osservato in questo studio di estensione di 24 mesi è sovrapponibile al profilo di sicurezza noto di Lucentis.
Nello studio di fase IIIb RETAIN, 372 pazienti con perdita dell'acuità visiva secondaria a DME sono stati randomizzati a ricevere le seguenti iniezioni intravitreali:
ranibizumab 0.5 mg con concomitante fotocoagulazione laser nell'ambito di un regime terapeutico flessibile del tipo «treat-and-extend» (TE) (n = 121).
ranibizumab 0.5 mg in monoterapia in regime TE (n = 128) oppure.
ranibizumab 0.5 mg in monoterapia in regime PRN (n = 123).
In tutti i gruppi, ranibizumab è stato somministrato sotto forma di iniezione intravitreale mensile fino a raggiungere una BCVA stabile per almeno tre valutazioni mensili consecutive. La fotocoagulazione laser è stata eseguita al basale nello stesso giorno in cui è stata somministrata la prima iniezione di ranibizumab e successivamente al bisogno secondo i criteri dello studio ETDRS. Nell'ambito del regime TE, ranibizumab è stato somministrato successivamente a intervalli di 2, massimo 3 mesi. Nell'ambito del regime PRN, la BCVA è stata valutata ogni mese e, se necessario, nella stessa sede si è proceduto alla somministrazione di ranibizumab. In tutti i gruppi, il trattamento mensile è stato ripreso in seguito a un calo della BCVA secondario a progressione del DME e proseguito fino a raggiungere di nuovo una BCVA stabile. Lo studio si è protratto per 24 mesi.
Nello studio RETAIN il numero medio di iniezioni somministrate è stato di 12.4 (12.0) nel gruppo ranibizumab + laser in regime TE, 12.8 (12.0) nel gruppo ranibizumab da solo in regime TE e 10.7 (10.0) nel gruppo ranibizumab in regime PRN. Dopo le 3 iniezioni iniziali, il numero di appuntamenti programmati per il trattamento è stato di 13 e 20, rispettivamente, per il regime TE e PRN. Con entrambi i regimi terapeutici, più del 70% dei pazienti è riuscito a mantenere la BCVA con una frequenza di visite ≥2 mesi. Ulteriori trattamenti con il laser non sono stati associati, nell'ambito del regime TE, con un più basso numero medio di iniezioni di ranibizumab.
I risultati sono riassunti nella tabella seguente:
Tabella 0-6 - Risultati dello studio RETAIN
Outcome vs. basale | Ranibizumab con regime TE | Ranibizumab con regime TE | Ranibizumab con regime PRN |
Variazione media della BCVA dal mese 1 al mese 12 (DS) | 5.9 (5.5)b | 6.1 (5.7)b | 6.2 (6.0) |
Variazione media della BCVA dal mese 1 al mese 24 (DS) | 6.8 (6.0) | 6.6 (7.1) | 7.0 (6.4) |
Variazione media della BCVA dopo 24 mesi (DS) | 8.3 (8.1) | 6.5 (10.9) | 8.1 (8.5) |
Aumento di ≥10 lettere e della BCVA ≥84 (%) dopo 24 mesi | 43.6 | 40.8 | 45.3 |
Aumento di ≥15 lettere e della BCVA ≥84 (%) dopo 24 mesi | 25.6 | 28.0 | 30.8 |
Calo di ≥10 lettere dopo 24 mesi | 2.6 | 7.2 | 3.4 |
Calo di ≥15 lettere dopo 24 mesi | 0.9 | 4.0 | 2.6 |
b p < 0.0001 | |||
Negli studi sul DME il miglioramento della BCVA è stato accompagnato in tutti i gruppi di trattamento da una graduale riduzione dello spessore retinico centrale (CRT) medio.
Il trattamento della retinopatia diabetica non proliferativa (NPDR) da moderata a grave o della retinopatia diabetica proliferativa (PDR)
La sicurezza e l'efficacia clinica di Lucentis in pazienti con retinopatia diabetica proliferativa (PDR) sono state valutate in uno studio di non inferiorità multicentrico, randomizzato, controllato in attivo in fase III a disegno parallelo (Protocol S) che ha coinvolto 305 pazienti (394 occhi di studio) con PDR con o senza DME (edema maculare diabetico) al basale. In questo studio sono stati confrontati 0.5 mg di ranibizumab iniettato per via intravitreale con il trattamento standard con fotocoagulazione panretinale (PRP). Un totale di 191 occhi (48.5%) sono stati randomizzati e assegnati al trattamento con 0.5 mg di ranibizumab e 203 occhi sono stati randomizzati (51.5%) e assegnati al trattamento con PRP. Un totale di 88 occhi (22.3%) presentava DME al basale: 42 (22.0%) e 46 (22.7%) occhi nei gruppi ranibizumab e PRP, rispettivamente. Un totale di 306 occhi (77.7%) non presentava DME al basale: 149 (78.0%) e 157 (77.3%) occhi nei gruppi ranibizumab e PRP, rispettivamente.
Dopo 2 anni di trattamento, il punteggio della BCVA (best corrected visual acuity) era cambiato di +2.7 lettere dal basale nel gruppo Lucentis e di -0.7 lettere dal basale nel gruppo PRP. La differenza di 3.5 lettere rientrava nel margine di non inferiorità, confermando così la non inferiorità di Lucentis rispetto al PRP.
La variazione di gravità della retinopatia diabetica è stata valutata utilizzando fotografie del fondo oculare con il punteggio di gravità della retinopatia diabetica (DRSS) dallo Early Diabetic Retinopathy Study (ETDRS). In questo studio, il 41.8% degli occhi trattati con ranibizumab (n = 189) ha mostrato un miglioramento della DRSS di almeno 2 livelli al mese 12, rispetto al 14.6% degli occhi trattati con PRP (n = 199).
In una meta-analisi di 3 studi di fase III randomizzati, in doppio cieco e controllati in attivo [D2301 (RESTORE), D2303 (REVEAL) e D2305 (REFINE)] eseguiti su un totale di 875 pazienti con DME, il 48.4% dei 315 pazienti trattati con ranibizumab con NPDR da moderata a grave o PDR (n = 192) ha mostrato un miglioramento della DRSS di almeno 2 livelli al mese 12, rispetto al 14.6% dei pazienti trattati con laser (n = 123).
Trattamento della perdita di acuità visiva causata da edema maculare secondario a RVO
La sicurezza e l'efficacia clinica di Lucentis in pazienti con perdita dell'acuità visiva causata da edema maculare secondario a occlusione venosa retinica (RVO) sono state valutate nei due studi randomizzati, in doppio cieco, controllati BRAVO (n = 397) e CRUISE (n = 392). In entrambi gli studi, i pazienti hanno ricevuto 0.3 mg o 0.5 mg di ranibizumab o trattamento sham. Nello studio BRAVO, la fotocoagulazione laser è stata ammessa come trattamento di salvataggio per tutti i pazienti in qualunque momento dello studio a partire dal terzo mese. I risultati degli studi BRAVO e CRUISE sono riportati sinteticamente nelle tabelle seguenti.
Tabella 0-7 - Studio BRAVO: risultati al mese 6 e 12
Variazione dell'acuità visiva | Trattamento sham | Ranibizumab 0.5 mg |
Variazione media in lettere della migliore acuità visiva corretta al mese 6 vs. l'acuità visiva all'inizio dello studioa | +7.3 | +18.3 |
Variazione media in lettere della migliore acuità visiva corretta al mese 12 vs. l'acuità visiva all'inizio dello studio | +12.1 | +18.3 |
Percentuale di pazienti con aumento dell'acuità visiva ≥15 lettere (%) al mese 6 | 28.8% | 61.1% |
Percentuale di pazienti con aumento dell'acuità visiva ≥15 lettere (%) al mese 12 | 43.9% | 60.3% |
Percentuale di pazienti con trattamento di salvataggio con laser oltre 12 mesi | 61.4% | 34.4% |
a:p < 0.0001 | ||
Tabella 0-8 - Studio CRUISE: risultati al mese 6 e 12
Variazione dell'acuità visiva | Trattamento sham (n = 132) | Ranibizumab 0.5 mg (n = 131) |
Variazione media in lettere della migliore acuità visiva corretta al mese 6 vs. l'acuità visiva all'inizio dello studioa | +0.8 | +14.9 |
Variazione media in lettere della migliore acuità visiva corretta al mese 12 vs. l'acuità visiva all'inizio dello studio | +7.3 | +13.9 |
Percentuale di pazienti con aumento dell'acuità visiva ≥15 lettere (%) al mese 6 | 16.9% | 47.7% |
Percentuale di pazienti con aumento dell'acuità visiva ≥15 lettere (%) al mese 12 | 33.1% | 50.8% |
a p < 0.0001 | ||
In entrambi gli studi (BRAVO e CRUISE), i pazienti trattati con ranibizumab hanno evidenziato una riduzione continua dello spessore retinico centrale.
Dopo il trattamento con ranibizumab al mese 6 e 12, oltre alla funzione visiva è migliorata anche la qualità di vita dei pazienti, misurata sulla base del punteggio al questionario sulla funzione visiva del National Eye Institute (VFQ‑25). La differenza tra il gruppo trattato con ranibizumab 0.5 mg e il gruppo di controllo al mese 6 è stata accertata con valori di p compresi tra 0.02 e 0.0002.
Dopo 12 mesi, i risultati ottenuti dallo studio di estensione HORIZON rispetto agli studi BRAVO e CRUISE hanno evidenziato quanto segue:
la ridotta frequenza dei trattamenti scelta nello studio HORIZON ha influito in misura minima sui pazienti affetti da BRVO, i quali hanno mantenuto l'iniziale miglioramento della funzione visiva osservato nello studio BRAVO (+17.5 lettere dopo 24 mesi con un dosaggio di 0.5 mg e un numero medio di 2.4 iniezioni al secondo anno).
Al contrario, la ridotta frequenza dei trattamenti stabilita per i pazienti affetti da CRVO è stata associata alla riduzione dell'acuità visiva raggiunta nello studio CRUISE (+12 lettere dopo 24 mesi con un dosaggio di 0.5 mg e un numero medio di 3.8 iniezioni al secondo anno).
Studio E2401 (CRYSTAL) e studio E2402 (BRIGHTER)
La sicurezza e l'efficacia clinica di Lucentis per 24 mesi sono state valutate nell'ambito degli studi BRIGHTER e CRYSTAL su pazienti con disturbi della vista dovuti a edema maculare secondario a RVO. Per gli studi sono stati reclutati pazienti con BRVO (n = 455) e CRVO (n = 357). In entrambi gli studi, i pazienti hanno ricevuto ranibizumab 0.5 mg al bisogno. BRIGHTER era uno studio a 3 bracci randomizzato, con controllo attivo che ha confrontato ranibizumab 0.5 mg somministrato in monoterapia o in combinazione con la fotocoagulazione laser verso la sola fotocoagulazione laser. Dopo 6 mesi, i pazienti inclusi nel braccio di trattamento con la sola fotocoagulazione laser hanno potuto ricevere ranibizumab 0.5 mg. CRYSTAL è uno studio a braccio singolo su ranibizumab 0.5 mg in monoterapia.
I risultati funzionali e anatomici più importanti ottenuti dagli studi BRIGHTER e CRYSTAL sono riportati nella tabella 9 e illustrati nelle figure 1‑0 e 2‑0.
Tabella 0-9 - Risultati al mese 6 (BRIGHTER) e al mese 24 (BRIGHTER e CRYSTAL)
BRIGHTER | CRYSTAL | |||
Lucentis 0.5 mg | Lucentis 0.5 mg + laser | Laser* | Lucentis 0.5 mg | |
Variazione media della BCVA al mese 6b (lettere) (DS) | +14.8 (10.7) | +14.8 (11.13) | +6.0 (14.27) | +12.0 (13.95) |
Variazione media della BCVA al mese 24b (lettere) (DS) | +15.5 (13.91) | +17.3 (12.61) | +11.6 (16.09) | +12.1 (18.60) |
Percentuale di pazienti che ha ottenuto un aumento ≥15 lettere della BCVA al mese 24 | 52.8% | 59.6% | 43.3% | 49.2% |
Numero medio di iniezioni (DS) (mesi 0–23) | 11.4 (5.81) | 11.3 (6.02) | N/A | 13.1 (6.39) |
* A partire dal mese 6 è stato permesso il trattamento con ranibizumab 0.5 mg (24 pazienti sono stati trattati solo con il laser). b p < 0.0001 per entrambi i confronti oggetto dello studio BRIGHTER al mese 6: Lucentis 0.5 mg vs. laser e Lucentis 0.5 mg + laser vs laser. | ||||
Figura 0-1 - BRIGHTER: variazione media della BCVA vs. basale in un periodo di 24 mesi

Figura 0-2 - CRYSTAL: variazione media della BCVA vs. basale in un periodo di 24 mesi

Il miglioramento dell'acuità visiva è risultato simile con o senza ischemia retinica. Nello studio BRIGHTER, i pazienti con o senza ischemia retinica (rispettivamente n = 87 e n = 35), che sono stati trattati con ranibizumab in monoterapia, al mese 24, hanno evidenziato una variazione media rispetto al basale di +15.4 e +12.9 lettere, rispettivamente. Nello studio CRYSTAL, i pazienti con o senza ischemia retinica (rispettivamente n = 107 e n = 109) hanno evidenziato una variazione media rispetto al basale di +11.1 e +12.9 lettere, rispettivamente.
Nei pazienti con una durata della malattia < 3 mesi, gli studi BRIGHTER e CRYSTAL hanno riscontrato un miglioramento dell'acuità visiva al mese 1 di 13.3 e 10.0 lettere, rispettivamente, e al mese 24 di 17.7 e 13.2 lettere, rispettivamente. Quando la durata della malattia è stata ≥12 mesi, il miglioramento dell'acuità visiva è stato al mese 1 di 4.8 e 7.9 lettere, rispettivamente, e al mese 24 di 8.4 e 8.6 lettere, rispettivamente. Si deve considerare di iniziare il trattamento fin dal momento della diagnosi.
Il profilo di sicurezza di ranibizumab osservato negli studi di 24 mesi è coerente con il profilo di sicurezza di Lucentis emerso dagli studi precedenti.
Trattamento della perdita dell'acuità visiva causata da CNV–studio G2301 (MINERVA)
La sicurezza e l'efficacia clinica di Lucentis in pazienti con perdita dell'acuità visiva causata da CNV secondaria a eziologie diverse rispetto ad AMD neovascolare e PM, sono state valutate sulla base dei dati a 12 mesi ottenuti dallo studio randomizzato, in doppio cieco, controllato con trattamento sham G2301 (MINERVA). Per l'analisi sono stati predefiniti cinque sottogruppi di pazienti, suddivisi per eziologia (strie angioidi, retinocoroidopatia postinfiammatoria, corioretinopatia sierosa centrale, corioretinopatia idiopatica e altre eziologie). 178 pazienti sono stati randomizzati in rapporto 2:1 a ricevere:
ranibizumab 0.5 mg all'inizio dello studio, seguito da un trattamento basato su un regime personalizzato in funzione dell'attività della malattia;
iniezione sham all'inizio dello studio, seguita da un trattamento basato su un regime personalizzato in funzione dell'attività della malattia;
A partire dal mese 2, tutti i pazienti hanno ricevuto il trattamento con ranibizumab al bisogno. L'endpoint primario è stato la variazione della migliore acuità visiva corretta (BCVA) dal basale al mese 2.
I risultati principali dello studio MINERVA sono riportati sinteticamente nelle tabelle 10 e 11, come anche nella figura 3‑0.
Tabella 0-10 - Risultati a 2 mesi (MINERVA)
Ranibizumab 0.5 mg | Trattamento sham | |
|---|---|---|
Variazione media della BCVA dal basale al mese 2 (lettere) (valore medio secondo il metodo dei minimi quadrati) a | + 9.5 | -0.4 |
Percentuale di pazienti con un aumento di ≥15 lettere dal basale o con un valore di 84 lettere al mese 2 | 31.4% | 12.3% |
Percentuale di pazienti che dal basale al mese 2 non hanno perso più di 15 lettere | 99.2% | 94.7% |
Diminuzione dello spessore foveale medio dal basale al mese 2 (valore medio secondo il metodo dei minimi quadrati) a | 77 µm | -9.8 µm |
a confronto unilaterale (p < 0.001) con controllo sham | ||
Figura 0-3 - Variazione media della BCVA dal basale al mese 12 nel tempo (MINERVA)

Dal confronto tra ranibizumab e le iniezioni sham al mese 2 è stato osservato un effetto coerente del trattamento sull'eziologia, e ciò sia sulla popolazione oggetto di studio in generale, sia nei singoli sottogruppi suddivisi per eziologia.
Tabella 0-11 - Effetto del trattamento sulla popolazione oggetto di studio in generale e sui sottogruppi definiti all'inizio dello studio in funzione dell'eziologia della CNV, per la variabile primaria al mese 2 (MINERVA)
Popolazione generale e suddivisa per eziologia all'inizio dello studio | Effetto del trattamento vs. sham (lettere) | Numero di pazienti |
|---|---|---|
Popolazione generale | 9.9 | 175* |
Strie angioidi | 14.6 | 27 |
Retinocoroidopatia postinfiammatoria | 6.5 | 27 |
Corioretinopatia sierosa centrale | 5.0 | 23 |
Corioretinopatia idiopatica | 11.4 | 62 |
Altre eziologiea | 10.6 | 36 |
a comprende eziologie della CNV che non rientrano negli altri sottogruppi
* Numero di pazienti con dati disponibili per l'analisi
Il miglioramento dell'acuità visiva è stato accompagnato da una diminuzione dello spessore foveale nell'arco di 12 mesi.
Il numero medio di iniezioni di ranibizumab somministrate nell'arco di 12 mesi nell'occhio oggetto di studio è stato di 5.8 nel gruppo trattato con ranibizumab e di 5.4 nel gruppo che trattato con iniezioni sham.
Bambini e adolescenti
Cinque adolescenti di età compresa tra 12 e 17 anni con perdita dell'acuità visiva causata da CNV (1 caso di CNV subfoveale in drusen del nervo ottico; 1 caso di CNV iuxtafoveale e 1 caso di CNV subfoveale in presenza di CNV idiopatica; 2 casi di CNV subfoveale in presenza di malattia di Best) hanno ricevuto un trattamento iniziale con ranibizumab 0.5 mg seguito da un regime terapeutico personalizzato in funzione dei segni di attività della malattia (ad es. compromissione dell'acuità visiva, permeabilità intra- e sottoretinica, emorragie o stravasi). La variazione della BCVA dal basale al mese 12 è migliorata in tutti e cinque i pazienti, passando da +5 a +38 lettere. Il miglioramento dell'acuità visiva è stato accompagnato da una stabilizzazione o diminuzione dello spessore foveale medio nell'arco di 12 mesi (ΔCSFT0-12 mesi: da -286 μm a +10 μm). Nei 12 mesi sono state somministrate da 2 a 5 iniezioni nell'occhio oggetto di studio (cfr. il capitolo «Posologia/impiego»).
Trattamento della perdita dell'acuità visiva causata da CNV secondaria a PM
La sicurezza e l'efficacia clinica di Lucentis in pazienti con perdita dell'acuità visiva causata da CNV secondaria a PM sono state valutate sulla base dei dati a 12 mesi ottenuti dallo studio cardine, controllato, in doppio cieco, randomizzato RADIANCE. La finalità di studio era di valutare due differenti regimi posologici di ranibizumab 0.5 mg per iniezione intravitreale rispetto alla PDT con verteporfina (vPDT, terapia fotodinamica con Visudyne).
I 277 pazienti sono stati randomizzati a uno dei seguenti bracci:
Gruppo I (ranibizumab 0.5 mg, regime posologico basato su criteri di stabilità definiti come nessun cambiamento della migliore acuità visiva corretta (BCVA) rispetto a due valutazioni mensili precedenti).
Gruppo II (ranibizumab 0.5 mg, regime posologico basato su criteri di attività della malattia definiti come perdita dell'acuità visiva per permeabilità intra- e sottoretinica o stravaso attivo causato da lesioni da CNV rilevate alla OTC e/o all'angiografia con fluoresceina).
Gruppo III (vPDT – i pazienti hanno potuto ricevere ranibizumab a partire dal mese 3).
Nei 12 mesi dello studio, i pazienti del gruppo I hanno ricevuto in media 4.6 iniezioni (da 1 a 11 iniezioni) e quelli del gruppo II 3.5 iniezioni (da 1 a 12 iniezioni). Nel gruppo II (in cui i pazienti hanno ricevuto la dose raccomandata in funzione dell'attività della malattia, cfr. il capitolo «Posologia/impiego»), al 50.9% dei pazienti sono state somministrate una o due iniezioni, al 34.5% da 3 a 5 iniezioni e al 14.7% da 6 a 12 iniezioni nell'arco dei 12 mesi dello studio. Il 62.9% dei pazienti del gruppo II non ha richiesto iniezioni durante la seconda metà dello studio.
I risultati più importanti ottenuti dallo studio RADIANCE sono illustrati sinteticamente nella tabella 12 e nella figura 4.
Tabella 0-12 - Risultati al mese 3 e al mese 12 (RADIANCE)
Gruppo I Ranibizumab 0.5 mg stabilità dell'acuità visiva (n = 105) | Gruppo II Ranibizumab 0.5 mg attività della malattia (n = 116) | Gruppo III vPDT* (n = 55) | |
|---|---|---|---|
Mese 3 | |||
| Variazione media della BCVA dal mese 1 al mese 3 vs. basalea (lettere) | +10.5 | +10.6 | +2.2 |
| Percentuale di pazienti con miglioramento della BCVA di | |||
| ≥10 lettere o con BCVA complessivamente di ≥84 lettere | 61.9% | 65.5% | 27.3% |
| ≥15 lettere o con BCVA complessivamente di ≥84 lettere | 38.1% | 43.1% | 14.5% |
Mese 12 | |||
| Numero di iniezioni fino al mese 12: | |||
| Media | 4.6 | 3.5 | N/A |
| Mediana | 4.0 | 2.0 | N/A |
| Variazione media della BCVA dal mese 1 al mese 12 vs. basale (lettere) | +12.8 | 12.5 | N/A |
| Percentuale di pazienti con miglioramento della BCVA di | |||
| ≥10 lettere o con BCVA complessivamente di ≥84 lettere | 69.5% | 69.0% | N/A |
| ≥15 lettere o con BCVA complessivamente di ≥84 lettere | 53.3% | 51.7 | N/A |
| * Controllo comparativo fino al mese 3. I pazienti randomizzati a ricevere la vPDT sono stati ammessi al trattamento con ranibizumab a partire dal mese 3 (nel gruppo III, 38 pazienti hanno ricevuto ranibizumab a partire dal mese 3): p <0.00001 per il confronto con il controllo con vPDT | |||
Figura 0-4 Variazione media della BCVA vs. BCVA basale nel tempo fino al mese 12 (RADIANCE)

BL = basale; SE = errore standard del valore medio.
Figura:
Miglioramento medio dell'acuità visiva vs. BL ± SE (lettere)
Gruppo I trattato con ranibizumab 0.5 mg in funzione della stabilizzazione (n = 105)
Gruppo II trattato con ranibizumab 0.5 mg in funzione dell'attività della malattia (n = 116)
Gruppo III trattato con PDT con Visudyne (n = 55)
I pazienti randomizzati al gruppo vPDT hanno potuto essere avviati al trattamento con ranibizumab a partire dal mese 3.
Il miglioramento dell'acuità visiva è stato accompagnato da una riduzione dello spessore retinico centrale.
Rispetto ai pazienti del gruppo trattato con la vPDT, i pazienti dei gruppi che hanno ricevuto ranibizumab hanno evidenziato un beneficio (p < 0.05), riferito soggettivamente da loro stessi, in termini di miglioramento sia del punteggio composito sia dei singoli punteggi (funzione visiva generale, attività da vicino, salute mentale e autonomia) del questionario NEI VFQ‑25.
Trattamento della ROP nei neonati prematuri: studio H2301 (RAINBOW)
La sicurezza e l'efficacia clinica di Lucentis 0.1 mg nel trattamento della ROP nei neonati prematuri è stata valutata utilizzando i dati semestrali dello studio di superiorità a gruppi paralleli H2301 (RAINBOW), randomizzato, aperto, a tre bracci, disegnato per valutare l'uso di ranibizumab nel dosaggio da 0.1 mg e da 0.2 mg, somministrato sotto forma di iniezioni intravitreali, rispetto alla terapia laser. I pazienti idonei dovevano mostrare uno dei seguenti risultati retinici in entrambi gli occhi:
- Zona I, stadio della malattia 1+, 2+, 3 o 3+
- Zona II, stadio della malattia 3+
- ROP aggressivo posteriore (AP‑ROP)
Per questo studio, 225 pazienti sono stati randomizzati in un rapporto 1:1:1 a ricevere ranibizumab 0.1 mg (n = 77) o 0.2 mg (n = 74) per via intravitreale o terapia laser (n = 74).
Il successo del trattamento, misurato dall'assenza di ROP attiva e dall'assenza di effetti strutturali avversi in entrambi gli occhi 24 settimane dopo il primo trattamento dello studio, è stato del 75% nel gruppo di ranibizumab 0.1 mg e del 66.2% nel gruppo di terapia laser. La maggior parte dei pazienti trattati con ranibizumab 0.1 mg (77.6%) ha ricevuto una singola iniezione per occhio.
Dal gruppo di trattamento con ranibizumab 0.1 mg, un numero inferiore di pazienti è passato a una modalità di trattamento diversa rispetto al gruppo di terapia laser (16.9% vs. 24.3%) a causa della mancanza di risposta. Gli effetti strutturali avversi sono stati meno frequenti nel gruppo di trattamento con ranibizumab 0.1 mg (5 pazienti, 6.7%) rispetto al gruppo di terapia laser (7 pazienti, 10.1%). Inoltre, il 75% dei pazienti ha raggiunto la risoluzione della malattia entro 8 giorni nel gruppo di trattamento di ranibizumab 0.1 mg rispetto ai 22.5 giorni del gruppo di terapia laser.
Farmacocinetica
Assorbimento
La somministrazione intravitreale mensile di Lucentis in pazienti con AMD neovascolare produce concentrazioni sieriche di ranibizumab generalmente basse, con livelli massimi (Cmax) nettamente al di sotto della concentrazione necessaria per inibire del 50% l'attività del VEGF (11–27 ng/ml al test di proliferazione cellulare).
Distribuzione
La concentrazione sierica massima (Cmax) oscilla in genere intorno a 0.46–1.76 ng/ml, la concentrazione sierica minima (Cmin) varia da 0.04 a 0.29 ng/l. La Cmax sierica è risultata proporzionale alla dose somministrata in un range da 0.05 a 1.0 mg/occhio. La concentrazione sierica rilevata nei pazienti con DME e RVO è risultata comparabile a quella osservata nei pazienti con AMD neovascolare.
Metabolismo
Non applicabile.
Eliminazione
In base alla concentrazione sierica di ranibizumab, l'emivita media di eliminazione di ranibizumab dal vitreo nei pazienti con AMD neurovascolare è di circa 9 giorni. Si ritiene che le concentrazioni di ranibizumab nel siero siano circa 90 000 volte più basse di quelle che si osservano nel vitreo.
Disturbi della funzionalità epatica
Non sono disponibili studi specifici.
Disturbi della funzionalità renale
Non sono stati condotti studi prospettici sulla farmacocinetica di Lucentis nei pazienti con insufficienza renale. In un'analisi di popolazione condotta sui dati farmacocinetici rilevati in pazienti con AMD neovascolare, si è rilevato che il 68% (n = 136/200) dei pazienti soffriva di insufficienza renale (46.5% lieve, 20% moderata e 15% grave). Sui pazienti con RVO oggetto dello studio, il 48.2% (n = 253/525) soffriva di insufficienza renale (36.4% lieve, 9.5% moderata e 2.3% grave). La riduzione della clearance sistemica di ranibizumab è risultata statisticamente non significativa.
Popolazione pediatrica (neonati prematuri con ROP)
Dopo la somministrazione intravitreale di ranibizumab 0.1 mg in ciascun occhio in neonati prematuri con ROP, la concentrazione sierica di ranibizumab era più alta rispetto ai pazienti adulti con AMD neovascolare in trattamento con 0.5 mg per occhio. Secondo un'analisi farmacocinetica di popolazione, le differenze della Cmax e dell'AUCinf erano circa 8 e 5 volte superiori, rispettivamente. L'emivita sistemica apparente è stata di circa 6 giorni. In questa analisi non è stata trovata alcuna correlazione tra la concentrazione sistemica di ranibizumab e quella di VEGF.
Dati preclinici
La somministrazione intravitreale bilaterale di ranibizumab a scimmie cynomolgus in dosi tra 0.25 mg/occhio e 2.0 mg/occhio una volta ogni 2 settimane per periodi fino a 26 settimane ha prodotto effetti oculari dose-dipendenti.
Nella camera anteriore è stato riscontrato un aumento dose-dipendente di «flare» e di cellule che ha raggiunto il picco 2 giorni dopo l'iniezione. L'intensità della risposta infiammatoria è diminuita generalmente con le iniezioni successive o nel periodo di convalescenza, ma non sempre è stata completamente reversibile. Nella camera posteriore sono stati rilevati, soprattutto dopo la prima somministrazione, emorragie retiniche perivenose focali reversibili, infiltrati cellulari nel vitreo e mosche volanti, tendenzialmente dose-dipendenti e generalmente persistenti fino alla fine del trattamento. Nell'arco di 26 settimane di studio, l'intensità dell'infiammazione del vitreo è aumentata di pari passo con il numero di iniezioni e il trattamento è stato quindi interrotto prima del tempo. Tuttavia, il riscontro è risultato reversibile dopo la fase di convalescenza. Su alcuni animali, dopo un periodo relativamente lungo di forte infiammazione, è stata osservata la formazione di cataratta, la qual cosa sta a indicare che le modificazioni del cristallino sono state correlate a una grave infiammazione. Dopo l'iniezione intravitreale è stato osservato un innalzamento transitorio della pressione intraoculare, indipendentemente dalla dose somministrata.
Le modificazioni microscopiche del tessuto oculare sono state tutte correlate all'infiammazione e non sono state considerate indicative di processi degenerativi a carico delle strutture oculari. In alcuni casi sono state osservate modificazioni infiammatorie granulomatose a livello della testa nel nervo ottico. Queste modificazioni del segmento posteriore sono diminuite, e in alcuni casi si sono risolte completamente, nella fase di convalescenza.
Dopo l'iniezione intravitreale, non sono stati rilevati segni di tossicità sistemica nelle concentrazioni sieriche che sono risultate oltre 100 volte più elevate di quelle che si registrano con il normale uso terapeutico nell'uomo. In tutti gli animali trattati non sono stati riscontrati anticorpi anti-ranibizumab, né nel siero né nel vitreo.
I dati preclinici coprono soltanto l'esposizione sistemica dopo la somministrazione intravitreale. Non sono disponibili dati di mutagenicità, cancerogenicità e immunotossicità.
Tossicità per la riproduzione
Nelle scimmie gravide, l'iniezione intravitreale di ranibizumab con un'esposizione sistemica massima pari a 0.9–7 volte l'esposizione clinica presumibilmente peggiore non ha avuto effetti embrio-fetotossici né teratogeni. Il trattamento non ha avuto ripercussioni nemmeno sul peso e sulla struttura della placenta. Ciononostante, per via degli effetti farmacologici, ranibizumab deve essere considerato potenzialmente teratogeno ed embrio-fetotossico.
L'assenza di effetti ranibizumab-mediati sullo sviluppo embrio-fetale è plausibilmente riconducibile soprattutto all'incapacità del frammento Fab di superare la barriera placentare. Tuttavia, è stato descritto un caso con alti livelli sierici materni di ranibizumab e presenza di ranibizumab nel siero fetale; se ne deduce che l'anticorpo anti-ranibizumab ha agito da proteina di trasporto (con regione FC) per ranibizumab, riducendone in tal modo la clearance nel siero materno e favorendo il passaggio attraverso la barriera placentare. Poiché gli studi sullo sviluppo embrio-fetale sono stati condotti su femmine gravide sane e la permeabilità placentare verso un frammento Fab potrebbe essere modificata da una malattia, i risultati dello studio devono essere interpretati con cautela.
Altre indicazioni
Incompatibilità
Non applicabile.
Poiché per questo medicamento non sono stati condotti studi di compatibilità, non lo si deve somministrare in combinazione con altri medicamenti.
Stabilità
Il medicamento non deve essere utilizzato oltre la data indicata con «EXP» sulla confezione.
Indicazioni particolari concernenti l'immagazzinamento
Conservare fuori dalla portata dei bambini.
Flaconcino con tappo perforabile: conservare il flaconcino nella scatola originale per proteggere il contenuto dalla luce. Conservare in frigorifero (2–8ºC). Non congelare.
Prima dell'uso, il flaconcino chiuso può essere conservato a temperatura ambiente (25ºC) per un massimo di 24 ore.
Siringa preriempita: conservare la siringa nel vassoio sigillato all'interno della scatola originale per proteggere il contenuto dalla luce. Conservare in frigorifero (2–8ºC). Non congelare.
Indicazioni per la manipolazione
Per ragioni microbiologiche, dopo l'apertura la soluzione iniettabile pronta all'uso di un flaconcino con tappo perforabile o la siringa preriempita deve essere utilizzata subito. Smaltire la soluzione inutilizzata (cfr. il capitolo «Posologia/impiego»). Il flaconcino e la siringa sono vanno usati per la somministrazione di una sola dose.
Flaconcino con tappo perforabile (adulti e neonati prematuri)
Il flaconcino è sterile. Non utilizzare il flaconcino se la confezione appare danneggiata. La sterilità del flaconcino può essere garantita solo se il sigillo della confezione è intatto. Non utilizzare il flaconcino se il colore o la trasparenza della soluzione appaiono alterati o se contiene particelle.
Per la preparazione e l'esecuzione dell'iniezione intravitreale sono necessari i seguenti componenti monouso:
- ago filtro da 5 μm (18G)
- siringa sterile da 1 ml
- ago per iniezione (13 mm, 30 gauge)
Questi componenti non sono inclusi nella confezione che contiene solo il flaconcino. La siringa sterile da 1 ml e l'ago per iniezione non sono inclusi nella confezione che contiene il flaconcino e l'ago filtro.
Siringa preriempita: prima dell'uso, la siringa chiusa di Lucentis può essere conservata a temperatura ambiente (25ºC) per un massimo di 24 ore.
Siringa preriempita (solo per adulti)
La siringa è monouso (cfr. il capitolo «Posologia/impiego»).
La siringa è sterile. Non utilizzare la siringa se la confezione appare danneggiata. La sterilità della siringa può essere garantita solo se il sigillo della confezione è intatto. Non utilizzare la siringa se il colore o la trasparenza della soluzione appaiono alterati o se contiene particelle.
Per preparare Lucentis per l'iniezione intravitreale, seguire le seguenti istruzioni:
Titolo | Istruzioni | Grafico/figura |
|---|---|---|
Prima di utilizzare la siringa leggere attentamente tutte le istruzioni. La siringa è monouso. La siringa è sterile. Non utilizzare il prodotto se la confezione appare danneggiata. Nota: la dose deve essere impostata su 0.05 ml. | ||
Descrizione della siringa preriempita |
| |
Preparazione | 1. Assicurarsi che la confezione contenga:
2. Sollevare il coperchio del vassoio in cui si trova la siringa e rimuovere con attenzione la siringa in condizioni asettiche. |
|
Controllo della siringa | 3. Controllare che:
4. Se una delle suddette condizioni non si verifica, smaltire la siringa e usarne una nuova. |
|
Rimozione del cappuccio della siringa | 5. Staccare (senza torcere né ruotare, cfr. Figura 2) il cappuccio della siringa flettendolo lateralmente. 6. Smaltire il cappuccio della siringa (cfr. Figura 3). |
|
Inserimento dell'ago | 7. Inserire con forza un ago per iniezione sterile da 30G lungo circa 1.3 cm sulla siringa avvitandolo bene sull'adattatore Luer lock (cfr. Figura 4). 8. Rimuovere il cappuccio dell'ago tirandolo con cautela in direzione verticale (cfr. Figura 5). Nota: non pulire mai l'ago. |
|
Espulsione delle bolle d'aria | 9. Tenere la siringa in posizione verticale. 10. Se sono presenti bolle d'aria, picchiettare delicatamente la siringa con il dito fino a farle salire in superficie (cfr. Figura 6). |
|
Impostazione della dose | 11. Tenere la siringa a livello degli occhi e spingere delicatamente lo stantuffo finché il bordo inferiore della cupola del tappo di gomma sia allineato con la tacca di misurazione della dose (cfr. Figura 7).
Nota: lo stantuffo non è attaccato al tappo di gomma al fine di evitare che l'aria venga aspirata nella siringa. |
|
Iniezione | Eseguire l'iniezione in condizioni asettiche. 12. Inserire l'ago per iniezione 3.5–4.0 mm posteriormente al limbus, nella camera vitreale, evitando il meridiano orizzontale e dirigendo l'ago verso il centro del globo oculare. 13. Iniettare lentamente la soluzione fino a far toccare la parte superiore del tappo di gomma e il fondo della siringa per somministrare il volume di 0.05 ml. 14. Per le iniezioni successive scegliere un altro punto della sclera. 15. Dopo l'iniezione, non ricoprire l'ago con un cappuccio né staccarlo dalla siringa. Smaltire la siringa usata insieme con l'ago in un apposito contenitore per oggetti taglienti o in conformità alla normativa locale vigente. |
|






Numero dell'omologazione
57664, 63277 (Swissmedic).
Titolare dell’omologazione
Novartis Pharma Schweiz AG, Risch; Domicilio 6343 Rotkreuz.
Stato dell'informazione
Ottobre 2020.
Composition
Principes actifs
Ranibizumabum.
Excipients
α,α-trehalosum dihydricum, Histidinum, Histidinum hydrochloricum monohydricum, Polysorbatum 20, Aqua ad iniect.
Forme pharmaceutique et quantité de principe actif par unité
Flacon: 10 mg/ml de ranibizumab (flacon contenant 2,3 mg de ranibizumab dans 0,23 ml de solution).
Seringue prête à l'emploi: 10 mg/ml de ranibizumab (seringue prête à l'emploi contenant 1,65 mg de ranibizumab dans 0,165 ml de solution).
Indications/Possibilités d’emploi
Lucentis est indiqué chez les adultes dans le traitement:
- de la forme exsudative (humide) de la dégénérescence maculaire liée à l'âge (DMLA humide),
- d'une perte de vision due à un œdème maculaire diabétique (OMD),
- de la rétinopathie diabétique non proliférante (RDNP) moyennement sévère à sévère ou de la rétinopathie diabétique proliférante (RDP),
- d'une perte de vision due à un œdème maculaire consécutif à une occlusion de veine rétinienne (occlusion de branche veineuse rétinienne OBVR et occlusion de la veine centrale de la rétine OVCR),
- d'une néovascularisation choroïdienne (NVC) active entraînant une baisse de vision,
- d'une perte de vision due à une néovascularisation choroïdienne (NVC) consécutive à une myopie pathologique (MP).
Lucentis est indiqué chez les prématurés dans le traitement de la rétinopathie du prématuré (RDP):
- RDP dans la zone I (stade de la maladie 1+, 2+, 3 ou 3+), zone II (stade de la maladie 3+) ou AP-RDP (RDP agressive postérieure).
Posologie/Mode d’emploi
Lucentis ne doit être utilisé que par un ophtalmologue qualifié disposant d'une infrastructure adéquate. Lucentis est injecté dans le corps vitré (voie intravitréenne). Un flacon de Lucentis (pour adultes et prématurés) contient 0,23 ml et une seringue prête à l'emploi (pour adultes uniquement) contient 0,165 ml. Les deux formes sont destinées à l'usage unique pour un seul patient.
Posologie usuelle
Posologie chez l'adulte
La dose recommandée de 0,5 mg est administrée sous forme d'injection intravitréenne unique. Cela correspond à un volume d'injection de 0,05 ml. L'intervalle entre deux injections dans le même œil ne doit pas être inférieur à un mois.
Le traitement est initié avec une injection par mois jusqu'à ce que l'acuité visuelle maximale soit atteinte et/ou jusqu'à l'absence de signe d'activité de la maladie. Chez les patients atteints de DMLA humide, d'OMD, de RDNP moyennement sévère à sévère ou de RDP, d'OBVR ou d'OVCR, trois injections mensuelles consécutives ou plus peuvent être nécessaires à l'initiation.
Ensuite, les intervalles de suivi et de traitement doivent être déterminés par le médecin. Le suivi de l'activité de la maladie peut inclure des examens cliniques, des contrôles de l'acuité visuelle et/ou des techniques d'imagerie, comme la tomographie à cohérence optique ou l'angiographie à la fluorescéine.
Les intervalles de traitement peuvent être allongés ou raccourcis progressivement en fonction des signes d'activité de la maladie et de l'évolution du traitement.
Si cela est indiqué selon l'avis du médecin, Lucentis peut aussi être utilisé mensuellement dans un œil donné.
Le traitement d'une perte de vision due à une NVC doit être déterminé pour chaque patient en fonction de l'activité de la maladie. Pour la NVC consécutive à une myopie pathologique (MP) ou pour d'autres NVC ne survenant pas dans le cadre d'une DMLA, on ne dispose d'aucune expérience ayant concerné une durée de traitement de plus d'une année.
Posologie chez le prématuré
La dose recommandée de Lucentis chez le prématuré est de 0,1 mg, administrée sous forme d'injection intravitréenne unique. Cela correspond à un volume d'injection de 0,01 ml. Chez le prématuré, le traitement de la rétinopathie du prématuré (RDP) commence par une dose unique, qui peut être administrée dans les deux yeux le même jour. Au total, jusqu'à trois injections peuvent être administrées dans chaque œil en six mois en présence de signes d'activité de la maladie. Si plusieurs injections sont nécessaires, l'intervalle entre deux injections doit être d'au moins un mois.
Patients présentant des troubles de la fonction hépatique
Aucune étude n'est disponible. Étant donné que la quantité de médicament atteignant la circulation systémique est négligeable, aucune mesure de précaution particulière n'est prévue.
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique n'est nécessaire pour les patients insuffisants rénaux (voir «Pharmacocinétique»).
Patients âgés (de plus de 65 ans)
Aucun ajustement posologique n'est nécessaire.
Enfants et adolescents
L'utilisation de Lucentis est déconseillée chez l'enfant et l'adolescent, car les données de sécurité et d'efficacité dans ce groupe de patients sont insuffisantes. Il existe des données limitées chez des patients adolescents âgés de 12 à 17 ans ayant une perte de vision due à une NVC (voir «Propriétés/Effets»).
Mode d'administration
L'administration intravitréenne doit être précédée d'une anamnèse approfondie recherchant d'éventuelles réactions d'hypersensibilité (voir «Mises en garde et précautions»).
L'administration de Lucentis doit avoir lieu sous des conditions d'asepsie (environnement approprié, recouvrement stérile, gants, ustensiles). Une anesthésie adaptée et un microbicide à large spectre topique pour la désinfection de la peau périoculaire, de la paupière et de la surface oculaire doivent être utilisés avant l'injection.
Prélever la totalité du contenu du flacon avec une aiguille avec filtre de 5 µm montée sur une seringue. Avant l'injection intravitréenne, retirer l'aiguille filtre et la remplacer par une aiguille à injection (13 mm, 30 Gauge). Expulser le contenu jusqu'à ce que l'extrémité du piston atteigne la graduation 0,05 ml (50 µl) sur la seringue. Le contenu d'un flacon est destiné à l'administration d'une dose unique. Jeter la solution non utilisée.
Chez l'adulte, l'aiguille à injection doit être introduite complètement, 3,5–4,0 mm derrière le limbe, en évitant le méridien horizontal et en visant le centre du globe oculaire.
Délivrer alors lentement le volume d'injection; changer de site sur la sclérotique lors des injections suivantes.
Après l'injection, surveiller la pression intraoculaire du patient. La surveillance doit comprendre le contrôle de la circulation sanguine de la papille du nerf optique immédiatement après l'injection, une mesure de la pression intraoculaire dans les 30 minutes, un examen ophtalmoscopique, un examen à la lampe à fente et un examen du fond d'œil après 2–7 jours. Instruire le patient pour qu'il signale immédiatement au médecin les signes éventuels d'une endophtalmie (voir «Mises en garde et précautions»).
Chez le prématuré, l'aiguille à injection doit être introduite 1,0 à 2,0 mm derrière le limbe, en direction du nerf optique. Le volume d'injection de 0,01 ml est ensuite administré.
Contre-indications
Hypersensibilité au ranibizumab ou à l'un des excipients. Lucentis est contre-indiqué chez les patients atteints d'infections oculaires ou périoculaires, ainsi que chez les patients présentant une inflammation intraoculaire active.
Mises en garde et précautions
Réactions liées aux injections intravitréennes
Une endophtalmie infectieuse et des décollements de la rétine peuvent survenir lors d'injections intravitréennes. C'est pourquoi il faudra utiliser des techniques d'injection aseptiques lors de l'injection de Lucentis. En outre, il faudra surveiller les patients dans les jours qui suivent l'injection afin de déceler une infection à temps et de pouvoir la traiter (voir «Effets indésirables»).
Chez l'adulte, une augmentation transitoire de la pression intraoculaire a été observée dans les 60 minutes suivant l'injection de Lucentis. Une augmentation prolongée de la pression intraoculaire a aussi été rapportée. C'est pourquoi il faudra surveiller la pression intraoculaire ainsi que la perfusion de l'artère centrale de la rétine et les traiter le cas échéant (voir «Effets indésirables»).
Traitement bilatéral
La sécurité d'emploi et l'efficacité d'un traitement simultané des deux yeux par Lucentis n'ont pas fait l'objet d'études spécialement conçues à cet effet.
Les données limitées à disposition concernant l'utilisation bilatérale de Lucentis (y compris administration le même jour) ne semblent pas indiquer un risque accru d'événement indésirable systémique par rapport au traitement unilatéral.
Événements thromboemboliques artériels
Il existe un risque potentiel d'événements thromboemboliques artériels dans le cas d'une application intravitréenne d'inhibiteurs du VEGF (vascular endothelial growth factor). Le risque est éventuellement accru chez les patients présentant un risque connu d'AVC (p.ex. ayant des antécédents d'AVC ou d'accident ischémique transitoire).
Immunogénicité
Dans tous les groupes de traitement, 0‑3% des patients qui n'étaient pas encore traités ont montré une réaction immunitaire à Lucentis. Des titres d'anticorps bas ont été observés chez 1–6% des patients après 12–24 mois lors d'une application mensuelle. Ces données sur l'immunogénicité reflètent le pourcentage des patients pour qui un test par électrochemiluminescence a donné des résultats positifs. Les données dépendaient fortement de la sensitivité et la spécificité du test. La signification clinique de la réaction immunitaire à Lucentis est actuellement incertaine. Iritis et inflammation du vitré ont été observées chez les patients montrant les plus hauts titres d'immunoréactivité.
Populations de patients chez lesquelles les données sont limitées
L'emploi de Lucentis chez des patients avec infection systémique active ou autres affections oculaires, telles que décollement de rétine ou trou maculaire n'a pas été étudié jusqu'ici.
Lucentis et photocoagulation au laser
Tant l'administration de Lucentis à des patients ayant subi dans le passé une photocoagulation au laser que l'emploi simultané de Lucentis et d'une photocoagulation au laser ont été étudiés. Si une injection de Lucentis est prévue le même jour qu'une photocoagulation au laser, l'injection devra être faite au plus tôt 30 minutes après le traitement au laser.
Durée du traitement
Pour la NVC consécutive à une myopie pathologique (MP) ou pour d'autres NVC ne survenant pas dans le cadre d'une DMLA, on ne dispose d'aucune expérience ayant concerné une durée de traitement de plus d'un an.
Interactions
Aucune étude spéciale d'interactions n'a été effectuée.
Grossesse/Allaitement
Grossesse
Il n'existe pas de données concernant l'emploi du ranibizumab chez la femme enceinte.
Des études chez le singe Cynomolgus n'ont révélé aucune toxicité directe ou indirecte ayant une incidence sur la grossesse, le développement embryonnaire ou le développement fœtal (voir «Données précliniques»). Le ranibizumab inhibe le VEGF‑A, un facteur angiogénique important pour la formation de néovaisseaux pendant le développement embryonnaire et fœtal et la formation du placenta. L'exposition systémique au ranibizumab est faible après administration oculaire. Compte tenu de son mécanisme d'action, le ranibizumab doit donc être considéré comme potentiellement tératogène et embryo-/fœtotoxique et ne doit pas être utilisé pendant la grossesse, sauf en cas de nécessité absolue. Chez les patientes qui envisagent une grossesse, le ranibizumab doit être arrêté 3 mois avant la conception.
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser des méthodes contraceptives efficaces pendant le traitement et les 30 jours suivants.
Allaitement
On ignore si Lucentis est excrété dans le lait maternel. Étant donné que de nombreuses substances sont excrétées dans le lait maternel et qu'une résorption et une perturbation de la croissance et du développement de l'enfant sont possibles, il est recommandé de ne pas allaiter pendant le traitement par Lucentis.
Fertilité
L'influence de Lucentis sur la fertilité masculine et féminine n'a pas été étudiée.
Effet sur l’aptitude à la conduite et l’utilisation de machines
Le traitement par Lucentis peut entraîner des troubles visuels passagers, ce qui peut altérer l'aptitude à la conduite et à l'utilisation de machines (voir «Effets indésirables»). Les patients présentant de tels signes ne doivent pas conduire de véhicules jusqu'à ce que ces troubles visuels passagers aient disparu.
Effets indésirables
Les 1315 patients au total ayant participé aux trois études cliniques de phase III sur le traitement de la DMLA humide fournissent la base des considérations en matière de sécurité. Tous les patients ont été traités 24 mois au minimum par Lucentis, et 440 d'entre eux ont reçu la dose recommandée de 0,5 mg.
Les effets indésirables graves liés à la procédure d'injection comprennent: endophtalmie, décollement de rétine rhegmatogène, déchirure de la rétine et cataracte traumatique iatrogène (voir «Mises en garde et précautions»).
Une inflammation intraoculaire et une hypertension intraoculaire ont également été observées chez les patients traités par Lucentis (voir «Mises en garde et précautions»).
Les effets indésirables mentionnés plus bas sont survenus durant les études contrôlées de phase III FVF2598g (MARINA), FVF2587g (ANCHOR) et FVF3192g (PIER) sur la DMLA humide de façon plus fréquente (au moins 2%) chez les patients avec DMLA humide traités par 0,5 mg de Lucentis que chez les patients du groupe témoin (traitement simulé (voir «Propriétés/Effets») ou PDT par la vertéporfine). Ils sont donc considérés comme des effets indésirables (EI) potentiels. Les données relatives à la sécurité spécifiées ci-dessous comprennent en outre tous les événements indésirables supposés avoir été au moins potentiellement causés par l'injection elle-même ou par le médicament, et qui sont survenus chez les 440 patients traités par 0,5 mg de Lucentis contre la DMLA humide dans le cadre du traitement combiné.
Collectif des patients atteints de RD
La sécurité de Lucentis a été examinée au cours d'une étude clinique de 24 mois dans l'étude de protocole S incluant entre autres 191 patients atteints de rétinopathie diabétique (RD) traités par le ranibizumab. Les événements oculaires et non oculaires observés étaient en accord avec ce à quoi on pourrait s'attendre dans le cas d'une population de patients diabétiques atteints de RD, ou ils étaient similaires aux événements ayant été observés lors d'études cliniques antérieures portant sur Lucentis en ce qui concerne leur fréquence et leur gravité.
Collectif des patients présentant une NVC
La sécurité de Lucentis a été examinée au cours d'une étude clinique de 12 mois (MINERVA) à laquelle ont participé 171 patients ayant une perte de vision due à une NVC et traités par le ranibizumab (voir «Propriétés/Effets»). Chez ces patients, le profil de sécurité a correspondu à celui observé dans des études cliniques antérieures menées avec Lucentis.
Collectif des patients atteints de NVC consécutive à une MP
La sécurité de Lucentis a été examinée au cours de l'étude clinique de 12 mois RADIANCE. Cette étude portait sur 244 patients présentant une NVC consécutive à une MP qui ont été traités par le ranibizumab (voir «Propriétés/Effets»). La fréquence et la sévérité des événements oculaires et non oculaires qui se sont produits au cours de cette étude ont été comparables à celles observées dans les études portant sur la DMLA humide.
Population atteinte de rétinopathie du prématuré (RDP)
- La sécurité de Lucentis 0,1 mg a fait l'objet d'une étude clinique de six mois (RAINBOW), portant sur 77 prématurés atteints de RDP et traités par Ranibizumab (voir «Propriétés/Effets»). Les effets indésirables oculaires observés lors de l'étude RAINBOW étaient conformes à ceux qui étaient également survenus chez des adultes pendant le traitement par Ranibizumab 0,5 mg ou qui correspondaient à un événement dans le cadre de la RDP. Les effets indésirables non oculaires dans cette étude clinique correspondaient généralement à ceux que l'on pouvait attendre chez des patients atteints de comorbidités multiples liées à la naissance prématurée. Pour des raisons théoriques, il convient de tenir compte du fait que la maturation d'autres organes pourrait être retardée après un traitement par des anti‑VEGF, ce qui pourrait entraîner des complications.
Fréquences: «très fréquents» (≥1/10), «fréquents» (≥1/100 à <1/10), «occasionnels» (≥1/1000 à <1/100), «rares» (≥1/10'000 à <1/1000), «très rares» (<1/10'000).
Infections et infestations
Très fréquents: rhinopharyngite (12,5–16,4%).
Fréquents: influenza, infections des voies urinaires.
Affections hématologiques et du système lymphatique
Fréquents: anémie.
Affections du système immunitaire
Fréquents: réactions d'hypersensibilité.
Affections psychiatriques
Fréquents: états anxieux.
Affections du système nerveux
Très fréquents: céphalées (8–15%).
Fréquents: accident vasculaire cérébral.
Affections oculaires
Très fréquents: inflammations intraoculaires (10–18%), inflammation du corps vitré (2,3–10%), décollement du corps vitré (18–19%), hémorragie rétinienne (25%), troubles visuels (6,6–10,5%), douleurs oculaires (27–32%), mouches volantes (7–25%), hémorragies conjonctivales (55–72%), irritation oculaire (12–15%), sensation de corps étranger dans les yeux (12–15%), larmoiement accru (10–14%), blépharite (8–11%), prurit (8,5–10%).
Fréquents: dégénérescence rétinienne, troubles de la rétine, décollement de la rétine, déchirures de la rétine, décollement de l'épithélium pigmentaire rétinien, déchirures dans l'épithélium pigmentaire rétinien, défauts visuels, hémorragies et altérations du corps vitré, uvéite, inflammation de l'iris, iridocyclite, cataracte (sous-capsulaire), opacification de la capsule postérieure du cristallin, kératite ponctuée, abrasions de la cornée, trouble de l'humeur aqueuse, vision trouble, saignements au site d'injection, hémorragie oculaire, conjonctivite (allergique), sécrétion oculaire, photopsie, photophobie, troubles oculaires, douleurs et œdème des paupières, hyperémie conjonctivale.
Occasionnels: endophtalmie, hypopion, hyphéma, kératopathie, adhérence de l'iris, nécrolyse et œdème de la cornée, stries dans la cornée, douleurs et irritations au site d'injection, cécité, irritations des paupières.
Affections cardiaques, affections vasculaires
Des événements thromboemboliques artériels selon la définition de l'Antiplatelet Trialists' Collaboration (1994), tels que des décès d'origine vasculaire, des infarctus du myocarde non mortels, des accidents vasculaires cérébraux ischémiques non mortels et des accidents vasculaires cérébraux hémorragiques non mortels, ont été mis en rapport avec la disponibilité systémique d'inhibiteurs très puissants du VEGF. Durant la première année, la proportion d'événements thromboemboliques s'est élevée à 2,3% dans les deux groupes de patients traités par Lucentis (0,3 et 0,5 mg). En comparaison, elle s'élevait à 1,3% dans le groupe témoin. Durant la deuxième année de l'étude MARINA, la proportion a atteint 3,0% pour les deux groupes de traitement, et 3,2% dans le groupe témoin.
Affections respiratoires, thoraciques et médiastinales
Fréquents: toux.
Affections gastro-intestinales
Fréquents: nausées.
Affections de la peau et du tissu sous-cutané
Occasionnels: réactions allergiques (rash, urticaires, prurit, érythème).
Affections musculosquelettiques et du tissu conjonctif
Très fréquents: douleurs articulaires (8–12%).
Investigations
Élévation de la pression intraoculaire.
Immunogénicité
Comme avec toutes les protéines thérapeutiques, il existe chez les patients traités par Lucentis un risque de réponse immunitaire. Les données d'immunogénicité reflètent le pourcentage de patients présentant un test positif à la recherche d'anticorps contre le ranibizumab par immuno-assay. Les résultats dépendaient beaucoup de la sensibilité et de la spécificité de l'assay.
Dans les études sur la DMLA, 0% à 3% des patients encore non traités de l'ensemble des groupes de traitement ont présenté une réponse immunitaire à Lucentis. Après des injections mensuelles de Lucentis durant 12 à 24 mois, environ 1% à 8% des patients avec DMLA néovasculaire étaient porteurs d'anticorps contre le ranibizumab.
Dans les études sur l'OMD, 0% à 2% des patients encore non traités de l'ensemble des groupes de traitement ont présenté une réponse immunitaire à Lucentis. Après des injections mensuelles de Lucentis durant 12 mois, environ 2% à 4% des patients avec OMD étaient porteurs d'anticorps contre le ranibizumab.
Dans les études sur l'OVR, 2% à 3% des patients encore non traités de l'ensemble des groupes de traitement ont présenté une réponse immunitaire à Lucentis. Après des injections mensuelles de Lucentis durant 12 mois, environ 4% à 5% des patients avec OVR étaient porteurs d'anticorps contre le ranibizumab.
La signification clinique de l'immunoréactivité à Lucentis n'est pas encore claire à ce jour.
Parmi les données agrégées des études cliniques en double aveugle et randomisées terminées, des infections/inflammations cutanées non oculaires et non graves se sont développées plus fréquemment chez les patients atteints d'OMD sous ranibizumab à 0,5 mg que chez les patients sous traitement de contrôle (1,85/100 années-patient par rapport à 0,27/100 années‑patient). Un lien avec le ranibizumab n'est jusqu'ici pas établi. Il existe toutefois un risque théorique d'un lien entre ces événements et l'inhibition du VEGF.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Surdosage
Des cas de surdosage non intentionnel (injection de volumes supérieurs à la dose recommandée de 0,05 ml de Lucentis) ont été rapportés lors des études cliniques sur la DMLA humide et durant la surveillance post-marketing.
Signes et symptômes
Les effets indésirables les plus fréquemment enregistrés étaient une élévation de la pression intraoculaire ainsi que des douleurs oculaires.
Traitement
En cas d'administration d'une dose trop élevée, il convient de surveiller la pression intraoculaire et, le cas échéant, de laisser le soin au médecin traitant d'instaurer le traitement adéquat.
Dans des études cliniques, des patients avec DMLA humide et OMD ont reçu des doses allant jusqu'à 2 mg de ranibizumab dans un volume d'injection de 0,05 ml à 0,10 ml. La nature et la fréquence des événements indésirables oculaires et systémiques correspondaient à celles rapportées pour la dose de 0,5 mg (dans 0,05 ml) de Lucentis.
Propriétés/Effets
Code ATC
S01LA04
Mécanisme d'action
Le principe actif de Lucentis (le ranibizumab) est un fragment d'anticorps monoclonal recombinant humanisé (Fab), dirigé contre le facteur de croissance vasculaire endothélial humain A (VEGF‑A). Le fragment se lie avec une grande affinité au VEGF-A et à ses isoformes. Les isoformes, tels que le VEGF121 et le VEGF165 sont formées par épissage alternatif de l'ARNm et l'isoforme VEGF110 est formée par protéolyse. La liaison du ranibizumab au VEGF‑A et à ses isoformes bloque l'activation des récepteurs VEGFR‑1 et VEGFR‑2 à la surface des cellules endothéliales.
Pharmacodynamique
L'activation des récepteurs VEGFR‑1 et VEGFR‑2 entraîne une prolifération des cellules endothéliales, une néovascularisation ainsi qu'une hyperperméabilité vasculaire. On suppose que tous ces facteurs contribuent à la progression de la forme néovasculaire de la dégénérescence maculaire liée à l'âge (DMLA), au développement de la NVC, y compris de la NVC consécutive à une myopie pathologique (MP), des œdèmes maculaires diabétiques (OMD) et des occlusions de veines rétiniennes (OVR) induisant la perte de vision.
Efficacité clinique
Traitement de la DMLA humide
La sécurité et l'efficacité cliniques du ranibizumab dans le traitement de la DMLA humide ont été étudiées dans trois études randomisées et en double aveugle, menées au total auprès de 1323 patients (Lucentis: N = 879, groupe témoin N = 444) avec DMLA néovasculaire. Les traitements simulés de l'étude MARINA et la PDT par Visudyne comme traitement actif de l'étude ANCHOR ont servi de bras de contrôle. Les patients inclus présentaient des lésions d'une taille allant jusqu'à 12 surfaces papillaires et une acuité visuelle dans l'œil étudié allant de 20/40 à 20/320 (selon Snellen). L'âge moyen des patients était de 77 ans. Dans le cadre des études cliniques, les patients ont reçu la consigne de s'appliquer eux-mêmes des gouttes antimicrobiennes dans les yeux (quatre fois par jour, 3 jours avant et après chaque injection).
L'étude MARINA de 24 mois a inclus 716 patients atteints de NVC classique minimale ou de NVC occulte. Ils ont reçu des injections intravitréennes mensuelles de 0,3 mg de Lucentis (N = 238), de 0,5 mg de Lucentis (N = 240) ou des injections simulées (N = 238). Durant les 24 mois de la phase de traitement, les patients ont reçu en moyenne 22 traitements sur les 24 possibles. Les résultats de l'étude MARINA observés après 12 mois de traitement ont été pour l'essentiel également confirmés après 24 mois de traitement (1 fois par mois) chez 90% des patients.
L'étude ANCHOR de 24 mois a inclus 423 patients atteints de NVC principalement classique. Ils ont reçu des injections intravitréennes mensuelles de 0,3 mg de Lucentis et une PDT simulée (N = 143), de 0,5 mg de Lucentis et une PDT simulée (N = 140) ou des injections intravitréennes simulées et une PDT active par la vertéporfine (N = 143). La première PDT simulée ou active par la vertéporfine a été administrée avec l'injection initiale de Lucentis. Ensuite, le rythme du traitement était tous les 3 mois lorsque l'œil étudié montrait une poursuite ou une reprise de l'hyperperméabilité vasculaire (angiographie fluorescéinique).
Les résultats sont résumés dans les tableaux suivants:
Tableau 0-1 Étude MARINA: résultats après 12 et 24 mois
Modification de l'acuité visuelle (en lettres, ETDRS) | Mois | Traitement simulé | Ranibizumab | Différence |
Perte de l'acuité visuelle <15 lettres (%)b | 12 mois | 62% | 95% | 32% |
24 mois | 53% | 90% | 37% | |
Gain de l'acuité visuelle ≥15 lettres (%)b | 12 mois | 5% | 34% | 29% |
24 mois | 4% | 33% | 29% | |
Modification moyenne de l'acuité visuelleb (lettres) | 12 mois | -10,5 (16,6) | +7,2 (14,4) | 17,5 |
24 mois | -14,9 (18,7) | +6,6 (16,5) | 21.1 | |
a après stratification b p <0,01 | ||||
Tableau 0-2 Étude ANCHOR: Résultats après 12 et 24 mois
Modification de l'acuité visuelle | Mois | PDT par vertéporfine | Ranibizumab 0,5 mg | Différence |
|---|---|---|---|---|
| Perte de l'acuité visuelle <15 lettres (%)b | 12 mois | 64% | 96% | 33% (25%, 41%) |
| 24 mois | 66% | 90% | 25% (16%, 34%) | |
| Gain de l'acuité visuelle ≥15 lettres (%)b | 12 mois | 6% | 40% | 35% (26%, 44%) |
| 24 mois | 6% | 41% | 35% (26%, 44%) | |
| Modification moyenne de l'acuité visuelleb(lettres) | 12 mois | -9,5 (16,4) | +11,3 (14,6) | 21,1 (17,5, 24,6) |
| 24 mois | -9,8 (16,4) | +10,7 (16,5) | 20,7 (16,8, 24,7) | |
a après stratification b p <0,01 | ||||
Dans les deux études MARINA et ANCHOR, l'amélioration de l'acuité visuelle observée avec Lucentis 0,5 mg à 12 mois s'est traduite par un bénéfice pour le patient. Ce bénéfice a été mesuré à l'aide des trois sous-échelles du National Eye Institute Visual Function Questionnaire (VFQ-25) qui étaient les critères secondaires pré-spécifiés pour l'évaluation de l'efficacité (activités liées à la vision de près, activités liées à la vision de loin et activités indépendantes de la vision). Toutes les différences entre Lucentis 0,5 mg et les deux groupes témoins ont été statistiquement significatives et cliniquement pertinentes, avec des valeurs de p comprises entre 0,009 et < 0,0001.
L'étude PIER a inclus 184 patients présentant des lésions NVC (avec ou sans composante classique). Ils ont reçu pendant les premiers 3 mois une injection intravitréenne par mois de Lucentis à 0,3 mg ou de Lucentis à 0,5 mg ou une injection intravitréenne simulée. D'autres injections de Lucentis ont eu lieu à intervalles de 3 mois. Après le mois 14, les patients qui avaient reçu une injection simulée pouvaient également être traités par Lucentis et à partir du mois 19, la fréquence des injections de Lucentis pouvait être augmentée. Les patients traités par Lucentis dans PIER ont reçu en moyenne 10 traitements en 24 mois.
Le critère principal d'évaluation de l'efficacité était la variation moyenne de l'acuité visuelle pendant les 12 mois. Après une augmentation initiale de l'acuité visuelle durant l'administration des doses mensuelles, les patients traités par une dose de Lucentis tous les 3 mois ont perdu de l'acuité visuelle, celle-ci revenant à la valeur initiale au mois 12 et cet effet a été conservé à 24 mois chez la plupart des patients traités par Lucentis (82%). Les données recueillies chez un nombre limité de patients ayant été passés à Lucentis après avoir reçu des injections simulées pendant plus d'un an indiquent qu'un début précoce du traitement est associé à une meilleure conservation de l'acuité visuelle.
Tableau 0-3 Étude PIER: résultats après 12 mois
Modification de l'acuité visuelle | Traitement simulé | Ranibizumab | Différence |
Perte de l'acuité visuelle <15 lettres (%)b | 49 | 90 | 37 |
Gain de l'acuité visuelle ≥15 lettres (%)b | 10 | 13 | 2 |
Modification moyenne de l'acuité visuelleb | -16,3 (22,3) | -0,2 (13,1) | 14,7 |
a après stratification b p < 0,0001 | |||
L'étude SAILOR de phase IIIb, multicentrique d'une durée de 1 an, a été réalisée chez des patients atteints de NVC due à une DMLA, aussi bien naïfs de traitement que prétraités. L'objectif primaire de cette étude était d'évaluer l'incidence des effets indésirables oculaires et non oculaires pendant les 12 mois de traitement. 2378 patients ont été randomisés dans 2 groupes qui ont reçu respectivement 0,3 mg ou 0,5 mg de Lucentis chaque mois pendant 3 mois; le traitement a ensuite été poursuivi en fonction des résultats, à intervalles d'au moins 1 mois.
Au total, il n'y a pas eu de déséquilibre entre les deux groupes quant aux effets indésirables oculaires et non oculaires. Il n'y a pas eu non plus de déséquilibre entre les deux groupes quant au nombre d'AVC. Sous 0,3 mg, 8 patients sur 1169 (0,7%, IC 95%: de 0,3% à 1,3%) en ont été atteints et sous 0,5 mg, 15 patients sur 1209 (1,2%, IC 95%: de 0,7% à 2%). Les patients ayant des facteurs de risque connus, tels qu'antécédents d'AVC ou d'accident ischémique transitoire, présentent vraisemblablement un risque accru d'AVC pendant le traitement par Lucentis.
Traitement de la perte de vision due à un OMD
L'efficacité et la sécurité cliniques de Lucentis chez les patients avec perte de vision due à un œdème maculaire diabétique (OMD) ont été étudiées dans le cadre de l'étude RESTORE, qui a inclus 345 patients présentant une perte de vision due à un OMD. L'étude comportait trois groupes: dans le groupe 1, les patients (N = 116) ont reçu initialement une injection intravitréenne de ranibizumab 0,5 mg en monothérapie et une photocoagulation au laser simulée. Les sujets du groupe 2 (N = 118) ont reçu initialement du ranibizumab 0,5 mg par injection intravitréenne et une photocoagulation au laser. Les patients du groupe 3 (N = 111) ont bénéficié initialement d'une photocoagulation au laser avec une injection simulée.
Le traitement par ranibizumab a ensuite été poursuivi sous forme d'injections intravitréennes mensuelles et interrompu en cas de vision stable sous Lucentis au cours de trois contrôles consécutifs. Après une interruption, le traitement a été repris en cas de détérioration de la vision du patient consécutive à la progression de l'OMD. Les répétitions des photocoagulations au laser ont été effectuées le même jour, au moins 30 minutes avant l'injection de ranibizumab, selon les critères de l'ETDRS.
Les résultats sont résumés dans les tableaux suivants:
Tableau 0-4 Étude RESTORE: Résultats à 12 mois
Changement de la meilleure vision corrigée | Ranibizumab | Ranibizumab | Laser | ||
Changement moyen de la meilleure acuité visuelle corrigée entre le 1er et le 12e mois en nombre de lettres, versus vision au début de l'étude (déviation standard)a | 6,1 (6,4) | 5,9 (7,9) | 0,8 (8,6) | ||
Changement moyen de la meilleure acuité visuelle corrigée au 12e mois en nombre de lettres, versus vision au début de l'étude (déviation standard) | 6,8 (8,3)a | 6,4 (11,8)b | 0,9 (11,4) | ||
Augmentation de la meilleure vision corrigée ≥10 lettres (% des patients) | 37,4c | 43,2 | 15,5 | ||
Augmentation de la meilleure vision corrigée ≥15 lettres (% des patients) | 22,6d | 22,9e | 8,2 | ||
a p <0,0001, b p = 0,0004, c p = 0,0001, d p = 0,0032, e p = 0,0021 | |||||
240 patients, qui avaient précédemment terminé l'étude RESTORE à 12 mois, ont été inclus dans l'étude d'extension de 24 mois multicentrique en ouvert (RESTORE Extension). Ils ont été traités par le ranibizumab 0,5 mg pro re nata (PRN) dans le même œil que celui sélectionné comme œil d'étude dans l'étude centrale. Le traitement était réinstauré mensuellement après diminution de la MAVC due à l'OMD et poursuivi jusqu'à ce qu'une stabilisation de la MAVC soit atteinte. De plus, un traitement par laser était réalisé conformément aux critères ETDRS, si l'investigateur le jugeait nécessaire.
Le nombre moyen d'injections administrées chez les patients traités par le ranibizumab dans l'étude centrale a été de 6,4 pendant la période d'extension de 24 mois. Parmi les 74 patients du bras traité par laser de l'étude centrale, 59 (79%) ont reçu le ranibizumab à un moment donné dans la phase d'extension. En moyenne, ces 59 patients ont reçu 8,1 injections de ranibizumab pendant la période d'extension de 24 mois. La proportion de patients qui n'a pas eu besoin d'un traitement par le ranibizumab durant la phase d'extension a été respectivement de 19%, 25% et 20% pour le groupe préalablement traité par ranibizumab, le groupe préalablement traité par ranibizumab + laser, et le groupe préalablement traité par laser seul.
Les résultats sont résumés dans le tableau suivant:
Tableau 0-5 Étude d'extension RESTORE: Résultats à 36 mois
Paramètre d'évaluation par rapport aux valeurs initiales dans l'étude centrale | Au préalable ranibizumab | Au préalable ranibizumab | Au préalable laser |
|---|---|---|---|
Variation moyenne de la MAVC par rapport aux valeurs initiales dans l'étude centrale au mois 36 (ET) | 8,0 (10,09) | 6,7 (9,59) | 6,0 (9,35) |
Gain ≥10 lettres par rapport aux valeurs initiales dans l'étude centrale ou MAVC ≥84 (%) au mois 36 | 39 (47,0) | 37 (44,6) | 31 (41,9) |
Gain ≥15 lettres par rapport aux valeurs initiales dans l'étude centrale ou MAVC ≥84 (%) au mois 36 | 23 (27,7) | 25 (30,1) | 16 (21,6) |
n = nombre de patients pour lesquels les valeurs étaient disponibles, non seulement au début de l'étude (étude centrale, mois 0) mais aussi à la visite du mois 36. * 59 (79%) des 74 patients avec traitement préalable par laser ont reçu du ranibizumab pendant la phase d'extension. | |||
Le profil de sécurité à long terme du ranibizumab observé dans l'étude d'extension de 24 mois est en accord avec le profil de sécurité connu de Lucentis.
Dans l'étude de phase IIIb (RETAIN), 372 patients présentant une baisse visuelle due à un OMD ont été randomisés pour recevoir des injections intravitréennes:
- de ranibizumab 0,5 mg avec une photocoagulation au laser concomitante selon un protocole «treat-and-extend» (TE) (n = 121),
- de ranibizumab 0,5 mg en monothérapie selon un protocole TE (n = 128) ou
- de ranibizumab 0,5 mg en monothérapie selon un protocole PRN (n = 123).
- Dans tous les groupes, le traitement par le ranibizumab a été initié par des injections mensuelles intravitréennes et poursuivi jusqu'à ce que la MAVC soit stable lors d'au moins trois évaluations mensuelles consécutives. La photocoagulation au laser a été réalisée au début de l'étude, le même jour que la première injection de ranibizumab, puis selon les besoins conformément aux critères ETDRS. Dans le protocole TE, le ranibizumab était ensuite administré à des intervalles prévus de 2 ou maximum 3 mois. Dans le protocole PRN, la MAVC était évaluée mensuellement et le ranibizumab était alors administré au cours de la même visite, si besoin. Dans tous les groupes, le traitement mensuel était réinstauré après une diminution de la MAVC due à la progression de l'OMD et poursuivi jusqu'à ce qu'une stabilisation de la MAVC soit de nouveau atteinte. La durée de l'étude a été de 24 mois.
- Dans l'étude RETAIN, le nombre moyen (médian) d'injections était de 12,4 (12,0) dans le groupe ranibizumab TE + laser, de 12,8 (12,0) dans le groupe ranibizumab TE seul, et de 10,7 (10,0) dans le groupe ranibizumab PRN. Après les trois premières visites mensuelles de traitement, le nombre de visites de traitement nécessaires dans le protocole TE était de 13 par rapport aux 20 requises dans le protocole PRN. Dans les deux schémas de traitement, plus de 70% des patients ont conservé leur MAVC à une fréquence de visite ≥2 mois. L'association du laser n'a pas diminué le nombre moyen d'injections de ranibizumab dans le protocole TE.
Les résultats sont résumés dans le tableau suivant:
Tableau 0-6 Résultats de l'étude RETAIN
Résultat exprimé par rapport aux valeurs initiales | TE Ranibizumab | TE Ranibizumab | PRN Ranibizumab |
Variation moyenne de la MAVC du mois 1 au mois 12 (ET) | 5,9 (5,5)b | 6,1 (5,7)b | 6,2 (6,0) |
Variation moyenne de la MAVC du mois 1 au mois 24 (ET) | 6,8 (6,0) | 6,6 (7,1) | 7,0 (6,4) |
Variation de la MAVC au mois 24 (ET) | 8,3 (8,1) | 6,5 (10,9) | 8,1 (8,5) |
Gain ≥10 lettres ou MAVC ≥84 (%) au mois 24 | 43,6 | 40,8 | 45,3 |
Gain ≥15 lettres ou MAVC ≥84 (%) au mois 24 | 25,6 | 28,0 | 30,8 |
Perte ≥10 lettres au mois 24 | 2,6 | 7,2 | 3,4 |
Perte ≥15 lettres au mois 24 | 0,9 | 4,0 | 2,6 |
b p <0,0001 | |||
Dans les études dans l'OMD, l'amélioration de la MAVC était accompagnée d'une réduction de la valeur moyenne de l'ECR au cours du temps dans tous les groupes de traitement.
Traitement de la rétinopathie diabétique non proliférante (RDNP) moyennement sévère à sévère ou de la rétinopathie diabétique proliférante (RDP)
La sécurité et l'efficacité cliniques de Lucentis chez les patients atteints de rétinopathie diabétique proliférante (RDP) ont été évaluées dans une étude de non-infériorité de phase III en design parallèle (protocole S), multicentrique, randomisée, contrôlée de manière active à laquelle 305 patients (394 yeux étudiés) atteints de RDP avec ou sans OMD (œdème maculaire diabétique) au point initial de l'étude ont participé et dans laquelle l'injection intravitréenne de 0,5 mg de ranibizumab a été comparée au traitement standard avec photocoagulation panrétinienne (PPR). Au total, 191 yeux (48,5%) ont été randomisés et traités par 0,5 mg de ranibizumab et 203 yeux (51,5%) ont été randomisés et traités par PPR. Au total, 88 yeux (22,3%) ont présenté un OMD au point initial: 42 (22,0%) et 46 (22,7%) des yeux dans le groupe avec le ranibizumab et la PPR respectivement. Au total, 306 yeux (77,7%) n'ont pas présenté d'OMD au point initial: 149 (78,0%) et 157 (77,3%) des yeux dans le groupe avec le ranibizumab et la PPR respectivement.
Après 2 ans de traitement, le score de MAVC a été modifié de +2,7 lettres depuis le point initial dans le groupe de Lucentis et de -0,7 lettres depuis le point initial dans le groupe de PPR. La différence de 3,5 lettres était comprise dans la marge de non-infériorité, de telle sorte que la non-infériorité de Lucentis par rapport à la PPR a été confirmée.
La modification du degré de gravité de la rétinopathie diabétique a été évaluée au moyen de photos du fond de l'œil par l'utilisation du score de gravité pour la rétinopathie diabétique (DRSS) de l'étude antérieure sur la rétinopathie diabétique (ETDRS). Dans cette étude, une amélioration du DRSS d'au moins 2 degrés avait eu lieu chez 41,8% des yeux sous traitement par le ranibizumab (n = 189) à 12 mois, comparé à 14,6% chez les yeux traités à la PPR (n = 199).
Dans une méta-analyse de 3 études de phase III randomisées, en double aveugle et contrôlées de manière active [D2301 (RESTORE), D2303 (REVEAL) et D2305 (REFINE)], réalisées chez 875 patients atteints d'OMD en tout, 48,4% des 315 patients atteints de RDNP moyenne à sévère ou de RDP (n = 192) traités par le ranibizumab ont montré une amélioration du DRSS d'au moins 2 degrés au 12e mois, comparé à 14,6% des patients traités au laser (n = 123).
Traitement de la perte de vision par œdème maculaire consécutif à une OVR
La sécurité et l'efficacité cliniques de Lucentis chez les patients avec perte de vision due à un œdème maculaire consécutif à l'occlusion d'une veine rétinienne (OVR) ont été étudiées au cours de deux études BRAVO (N = 397) et CRUISE (N = 392), randomisées et contrôlées en double aveugle. Dans les deux études, les patients ont reçu soit 0,3 mg de ranibizumab, soit 0,5 mg de ranibizumab, soit un traitement simulé. Dans BRAVO, une photocoagulation au laser était autorisée à tout moment de l'étude à partir du 3e mois dans tous les groupes, en guise de traitement de secours. Les résultats de BRAVO et CRUISE sont résumés dans les tableaux suivants.
Tableau 0-7 Étude BRAVO: Résultats à 6 mois et à 12 mois
Changement de l'acuité visuelle | Traitement simulé | Ranibizumab 0,5 mg |
Changement moyen de la meilleure | +7,3 | +18,3 |
Changement moyen de la meilleure vision corrigée au 12e mois en nombre de lettres versus vision au début de l'étude | +12,1 | +18,3 |
Proportion de patients avec un gain de l'acuité visuelle ≥15 lettres (%) au 6e mois | 28,8% | 61,1% |
Proportion de patients avec un gain de l'acuité visuelle ≥15 lettres (%) au 12e mois | 43,9% | 60,3% |
Proportion de patients ayant reçu un laser de secours pendant 12 mois | 61,4% | 34,4% |
a p <0,0001 | ||
Tableau 0-8 Étude CRUISE: Résultats à 6 mois et à 12 mois
Changement de l'acuité visuelle | Traitement simulé (n = 132) | Ranibizumab 0,5 mg (n = 131) |
Changement moyen de la meilleure vision corrigée au mois 6, en nombre de lettres, versus début de l'étudea | +0,8 | +14,9 |
Changement moyen de la meilleure vision corrigée au 12e mois, en nombre de lettres, versus début de l'étudea | +7,3 | +13,9 |
Proportion de patients avec un gain de l'acuité visuelle ≥15 lettres (%) au 6e mois | 16,9% | 47,7% |
Proportion de patients avec un gain de l'acuité visuelle ≥15 lettres (%) au 12e mois | 33,1% | 50,8% |
a p <0,0001 | ||
Les patients traités par ranibizumab ont présenté dans les deux études (BRAVO et CRUISE) une diminution constante de l'épaisseur centrale de la rétine.
Hormis l'amélioration de l'acuité visuelle sous ranibizumab aux mois 6 et 12, l'étude comportait également un volet d'évaluation de la qualité de vie à ces deux échéances, au moyen du National Eye Institute Visual Function Questionnaire (VFQ‑25). Les différences entre le groupe ranibizumab 0,5 mg et le groupe témoin ont donné des valeurs de p à 6 mois situées entre 0,02 et 0,0002.
Les résultats de l'étude d'extension HORIZON de BRAVO et CRUISE ont montré les éléments suivants après 12 mois:
La réduction de la fréquence des traitements dans l'étude HORIZON a eu peu de répercussion sur les patients OBVR qui ont conservé l'amélioration initiale de leur acuité visuelle telle qu'observée dans l'étude BRAVO (+17,5 lettres après 24 mois avec une dose de 0,5 mg et en moyenne 2,4 injections au cours de la deuxième année).
En revanche, la réduction de la fréquence des traitements chez les patients OVCR s'est traduite par une diminution de l'acuité visuelle acquise dans l'étude CRUISE (+12 lettres après 24 mois avec une dose de 0,5 mg et en moyenne 3,8 injections au cours de la deuxième année).
Études E2401 (CRYSTAL) et E2402 (BRIGHTER)
Les études BRIGHTER et CRYSTAL ont évalué la sécurité et l'efficacité cliniques de Lucentis sur 24 mois chez les patients atteints de troubles de la vision dus à un œdème maculaire consécutif à une OVR. Les études ont inclus des patients atteints d'OBVR (n = 455) et d'OVCR (n = 357). Dans les deux études, les patients ont reçu 0,5 mg de ranibizumab selon le besoin. BRIGHTER était une étude randomisée à 3 groupes, à contrôle par principe actif, dans laquelle le ranibizumab à 0,5 mg en monothérapie était comparé au ranibizumab en association avec la photocoagulation laser et à la photocoagulation laser seule. Après 6 mois, les participants du groupe laser en monothérapie ont pu recevoir 0,5 mg de ranibizumab. CRYSTAL était une étude à un groupe sur le ranibizumab à 0,5 mg en monothérapie.
Les principaux résultats fonctionnels et anatomiques des études BRIGHTER et CRYSTAL sont résumés dans le tableau 0‑9 et dans les figures 1‑0 et 2‑0.
Tableau 0-9 Résultats à 6 mois (BRIGHTER) et à 24 mois (BRIGHTER et CRYSTAL)
BRIGHTER | CRYSTAL | |||
Lucentis 0,5 mg | Lucentis 0,5 mg | Laser* | Lucentis 0,5 mg | |
Modification moyenne de la MAVC à 6 moisb (lettres) (ET) | +14,8 (10,7) | +14,8 (11,13) | +6,0 (14,27) | +12,0 (13,95) |
Modification moyenne de la MAVC à 24 moisb (lettres) (ET) | +15,5 (13,91) | +17,3 (12,61) | +11,6 (16,09) | +12,1 (18,60) |
Proportion de patients qui ont atteint une MAVC ≥15 lettres à 24 mois | 52,8% | 59,6% | 43,3% | 49,2% |
Nombre moyen d'injections (ET) (mois 0–23) | 11,4 (5,81) | 11,3 (6,02) | N/A | 13,1 (6,39) |
* À partir du 6e mois, un traitement avec ranibizumab 0,5 mg était autorisé (24 patients n'ont été traités que par laser). b: p <0,0001 pour les deux comparaisons dans l'étude BRIGHTER à 6 mois: Lucentis 0,5 mg comparé à laser et Lucentis 0,5 mg + laser comparé à laser. | ||||
Figure 1-0 BRIGHTER: Modification moyenne de la MAVC par rapport au niveau initial sur une période de 24 mois

Figure 2-0 CRYSTAL: Modification moyenne de la MAVC par rapport au niveau initial sur une période de 24 mois

L'amélioration de la vision s'est révélée similaire chez les patients avec et sans ischémie rétinienne: dans l'étude BRIGHTER, les patients avec ischémie rétinienne (n = 87) ou sans ischémie rétinienne (n = 35) traités par le ranibizumab en monothérapie présentaient à 24 mois, une modification moyenne par rapport au niveau initial de respectivement +15,4 et +12,9 lettres. Dans l'étude CRYSTAL, les patients avec ischémie rétinienne (n = 107) ou sans ischémie rétinienne (n = 109) présentaient une modification moyenne par rapport au niveau initial de respectivement +11,1 et +12,9 lettres.
Chez les patients malades depuis < 3 mois, une amélioration de l'acuité visuelle de respectivement 13,3 et 10,0 lettres a été observée à 1 mois pour les études BRIGHTER et CRYSTAL, et de respectivement 17,7 et 13,2 lettres à 24 mois. Pour une maladie présente depuis ≥12 mois, elle était respectivement de 4,8 et 7,9 lettres à 1 mois, et de respectivement 8,4 et 8,6 lettres à 24 mois. Une initiation du traitement lors du diagnostic est à envisager.
Le profil de sécurité du ranibizumab, observé dans ces études sur 24 mois, correspond au profil de sécurité de Lucentis observé dans les études précédentes.
Traitement d'une perte de vision due à une NVC–Étude G2301 (MINERVA)
La sécurité et l'efficacité clinique de Lucentis chez des patients ayant une perte de vision due à une NVC consécutive à des étiologies autres qu'une DMLA néovasculaire et une MP ont été évaluées sur la base des données à 12 mois de l'étude G2301 (MINERVA) randomisée, en double aveugle, contrôlée contre une injection simulée. Pour l'analyse, cinq sous-groupes ont été prédéfinis selon l'étiologie (stries angioïdes, choriorétinopathie post-inflammatoire, choriorétinopathie centrale séreuse, choriorétinopathie idiopathique et autres étiologies). 178 patients ont été randomisés selon un ratio 2:1 dans l'un des bras suivants:
Ranibizumab 0,5 mg au début de l'étude, puis schéma posologique individuel selon l'activité de la maladie.
Injection simulée au début de l'étude, puis schéma posologique individuel selon l'activité de la maladie.
À partir du mois 2, tous les patients ont reçu un traitement par le ranibizumab selon les besoins. Le critère d'évaluation principal était la variation de la meilleure acuité visuelle corrigée (MAVC) entre le début de l'étude et le mois 2.
Les principaux résultats de MINERVA sont résumés dans les tableaux 0‑10 et 0‑11 et dans la figure 3‑0.
Tableau 0-10 Résultats au mois 2 (MINERVA)
Ranibizumab 0,5 mg | Injections simulées | |
|---|---|---|
Variation moyenne de la MAVC entre le début de l'étude et le mois 2 (lettres) (moyenne des moindres carrés)a | +9,5 | -0,4 |
Proportion de patients ayant gagné ≥15 lettres depuis le début de l'étude ou ayant atteint 84 lettres au mois 2 | 31,4% | 12,3% |
Proportion de patients n'ayant pas perdu plus de 15 lettres entre le début de l'étude et le mois 2 | 99,2% | 94,7% |
Diminution de l'épaisseur fovéolaire centrale entre le début de l'étude et le mois 2 (moyenne des moindres carrés)a | 77 µm | -9,8 µm |
a Test unilatéral (p <0,001) avec comparaison avec les injections simulées | ||
Figure 3-0: Variation moyenne de la MAVC entre le début de l'étude et le mois 12 au cours du temps (MINERVA)

Lors de la comparaison du ranibizumab avec les injections simulées au mois 2, un effet cohérent a été observé, que ce soit dans la population globale de l'étude ou dans les différents sous-groupes définis selon l'étiologie.
Tableau 0-11 Efficacité du traitement pour la population globale et dans les sous-groupes définis selon l'étiologie au début de l'étude, pour la variable principale au mois 2 (MINERVA)
Population globale et sous-groupes stratifiés en fonction de l'étiologie au début de l'étude | Efficacité du traitement par rapport aux injections simulées (lettres) | Nombre de patients (n) (traitement + injections simulées) |
Population globale | 9,9 | 175* |
Stries angioïdes | 14,6 | 27 |
Choriorétinopathie post-inflammatoire | 6,5 | 27 |
Choriorétinopathie séreuse centrale | 5,0 | 23 |
Choriorétinopathie idiopathique | 11,4 | 62 |
Autres étiologiesa | 10,6 | 36 |
a Étiologies NVC non comprises dans les autres sous-groupes *Nombre de patients pour lesquels des données sont disponibles pour l'analyse | ||
L'amélioration de la vision s'est accompagnée d'une diminution de l'épaisseur fovéolaire centrale sur une période de 12 mois.
Le nombre moyen d'injections de ranibizumab sur une période de 12 mois dans l'œil étudié a été de 5,8 dans le bras ranibizumab et de 5,4 dans le groupe ayant reçu les injections simulées.
Enfants et adolescents
Cinq patients adolescents âgés de 12 à 17 ans ayant une perte de vision due à une NVC (1x NVC subfovéale lors de drusen papillaires; 1x NVC juxtafovéale et 1x NVC subfovéale lors d'une NVC idiopathique; 2x NVC subfovéale lors d'une maladie de Best) ont reçu un traitement initial par ranibizumab 0,5 mg, puis un schéma thérapeutique individualisé en fonction des signes d'activité de la maladie (p.ex. baisse de l'acuité visuelle, liquide intra/sous-rétinien, hémorragies ou fuites). La variation de la MAVC entre le début de l'étude et le mois 12 s'est améliorée chez les cinq patients et a atteint de + 5 à + 38 lettres. L'amélioration de la vision s'est accompagnée d'une stabilisation ou d'une diminution de l'épaisseur fovéolaire centrale pendant une période de 12 mois (ΔCSFT0‑12 mois: de -286 μm à +10 μm). 2 à 5 injections ont été administrées dans l'œil étudié pendant les 12 mois (voir «Posologie/Mode d'emploi»).
Traitement d'une perte de vision due à une NVC consécutive à une MP
La sécurité et l'efficacité cliniques de Lucentis chez les patients présentant une perte de vision due à une NVC consécutive à une MP ont été évaluées à partir des données portant sur une période de 12 mois provenant de l'étude pivot randomisée, contrôlée, et conduite en double aveugle RADIANCE. Cette étude a été conçue pour évaluer deux schémas posologiques de 0,5 mg de ranibizumab par injection intravitréenne en comparaison à une PDT à la vertéporfine (vPDT, thérapie photodynamique avec Visudyne).
Les 277 patients ont été randomisés dans l'un des bras d'étude suivants:
Groupe I (0,5 mg de ranibizumab, le schéma posologique s'est basé sur des critères de stabilité définis comme une absence de modification de la meilleure acuité visuelle corrigée (MAVC, best corrected visual acuity) par rapport à deux examens mensuels précédents);
Groupe II (0,5 mg de ranibizumab, le schéma posologique s'est basé sur des critères d'activité de la maladie définis comme une perte de vision en raison de la présence de liquide intra- ou sous-rétinien ou d'un écoulement actif de liquide suite à la lésion de NVC, avec mise en évidence par une TCO et/ou par une AF);
Groupe III (vPDT; les patients ont pu être traités par le ranibizumab dès le 3e mois).
Au cours des 12 mois de l'étude, les patients du groupe I ont reçu 4,6 injections en moyenne (entre 1 et 11 injections) et ceux du groupe II ont reçu 3,5 injections en moyenne (entre 1 et 12 injections). Dans le groupe aII (dans lequel les patients ont reçu le traitement recommandé sur la base de critères déterminés par l'activité de la maladie, voir «Posologie/Mode d'emploi»), 50,9% des patients ont nécessité une ou deux injections, 34,5% des patients ont nécessité trois à cinq injections et 14,7% des patients ont nécessité 6 à 12 injections au cours de la période d'étude de 12 mois. 62,9% des patients du groupe II n'ont pas nécessité d'injections supplémentaires au cours des six derniers mois de l'étude.
Les résultats thérapeutiques les plus importants de l'étude RADIANCE sont résumés dans le tableau 0‑12 et dans la figure 0‑4.
Tableau 0-12 Résultat du traitement au 3e mois et au 12e mois (RADIANCE)
Groupe I | Groupe II | Groupe III | |
|---|---|---|---|
3e mois | |||
Modification moyenne de la MAVC entre le 1er et le 3e mois par rapport au niveau de basea (lettres) | +10,5 | +10,6 | +2,2 |
Proportion des patients présentant une amélioration de la MAVC: |
|
|
|
de ≥10 lettres ou une MAVC globale ≥84 lettres | 61,9% | 65,5% | 27,3% |
de ≥15 lettres ou une MAVC globale ≥84 lettres | 38,1% | 43,1% | 14,5% |
12e mois | |||
Nombre d'injections jusqu'au 12e mois: |
|
|
|
Moyenne | 4,6 | 3,5 | N/A |
Médiane | 4,0 | 2,0 | N/A |
Modification moyenne de la MAVC entre le 1er et le 12e mois par rapport au niveau de base (lettres) | +12,8 | 12,5 | N/A |
Proportion des patients présentant une amélioration de la MAVC: |
|
|
|
de ≥10 lettres ou une MAVC globale ≥84 lettres | 69,5% | 69,0% | N/A |
de ≥15 lettres ou une MAVC globale ≥84 lettres | 53,3% | 51,7 | N/A |
* Contrôle comparatif jusqu'au 3e mois. Les patients randomisés dans le groupe vPDT ont pu recevoir un traitement par le ranibizumab dès le 3e mois (dans le groupe III, 38 patients ont reçu du ranibizumab dès le 3e mois): p< 0,00001 pour la comparaison au groupe témoin vPDT | |||
Figure 0-4 Modification moyenne de la MAVC par rapport à la MAVC initiale au cours du temps jusqu'au 12e mois (RADIANCE)

BL = niveau initial; ES = erreur standard de la moyenne.
Figure:
Amélioration moyenne de l'acuité visuelle par rapport au BL ± ES (lettres)
Groupe I avec 0,5 mg de ranibizumab après stabilisation (N = 105)
Groupe II avec 0,5 mg de ranibizumab après activité de la maladie (N = 116)
Groupe III avec PDT par Visudyne (N = 55)
Les patients qui ont été randomisés dans le groupe vPDT ont pu recevoir un traitement par le ranibizumab à partir du 3e mois.
L'amélioration de l'acuité visuelle a été accompagnée d'une diminution de l'épaisseur centrale de la rétine.
Dans les bras de traitement par le ranibizumab, un bénéfice subjectivement rapporté par les patients dans le questionnaire VFQ‑25 a été constaté par rapport au groupe vPDT (p < 0,05) en ce qui concerne l'amélioration du résultat combiné ainsi que du résultat dans plusieurs sous-échelles (vision globale, activités nécessitant la vision de près, condition psychique et statut d'indépendance fonctionnelle).
Traitement de la RDP chez le prématuré: étude H2301 (RAINBOW)
L'efficacité et la sécurité cliniques de Lucentis 0,1 mg dans le traitement de la RDP chez le prématuré ont été analysées dans le cadre de l'étude de supériorité randomisée, ouverte, à trois bras et en groupes parallèles H2301 (RAINBOW) de six mois, conçue pour évaluer l'utilisation du ranibizumab à des doses de 0,1 mg et 0,2 mg, administré par injection intravitréenne, en comparaison avec un traitement au laser. Les patients candidats devaient présenter l'un des signes rétiniens suivants dans les deux yeux:
- Zone I, stade de la maladie 1+, 2+, 3 ou 3+
- Zone II, stade de la maladie 3+
- RDP agressive postérieure (AP-RDP)
Dans le cadre de cette étude, 225 patients ont été randomisés selon un rapport 1:1:1 dans des groupes recevant le ranibizumab par voie intravitréenne à une dose de 0,1 mg (n = 77) ou de 0,2 mg (n = 74) ou un traitement au laser (n = 74).
La réussite du traitement, mesurée par l'absence de RDP active et d'effets indésirables structurels dans les deux yeux 24 semaines après le premier traitement de l'étude, était de 75% dans le groupe ranibizumab 0,1 mg et de 66,2% dans le groupe de traitement au laser. La majorité des patients traités par ranibizumab 0,1 mg (77,6%) ont reçu une seule injection dans chaque œil.
Dans le groupe de traitement ranibizumab 0,1 mg, moins de patients ont changé de mode de traitement en raison d'un manque de réponse que dans le groupe de traitement au laser (16,9% vs 24,3%). Les effets indésirables structurels ont été moins fréquents dans le groupe de traitement ranibizumab 0,1 mg (5 patients, 6,7%) que dans le groupe de traitement au laser (7 patients, 10,1%). En outre, 75% des patients ont obtenu une diminution de la maladie en 8 jours sous ranibizumab 0,1 mg, contre 22,5 jours dans le groupe de traitement au laser.
Pharmacocinétique
Absorption
L'administration intravitréenne mensuelle de Lucentis à des patients atteints de DMLA néovasculaire conduit à des concentrations sériques de ranibizumab généralement faibles et à une concentration sérique maximale (Cmax) nettement inférieure à la concentration ayant entraîné une inhibition de l'activité du VEGF de 50% (11–27 ng/ml dans un test in vitro sur la prolifération cellulaire).
Distribution
La concentration sérique maximale (Cmax) se situe généralement dans un intervalle compris entre 0,46 et 1,76 ng/ml et la concentration sérique minimale (Cmin) entre 0,04 et 0,29 ng/ml. La Cmax sérique était proportionnelle à la dose dans l'intervalle posologique compris entre 0,05 et 1,0 mg/œil. La concentration sérique chez les patients avec OMD et OVR était comparable à celle de patients avec DMLA néovasculaire.
Métabolisme
Non applicable.
Élimination
Sur la base de la concentration sérique de ranibizumab, la demi-vie d'élimination moyenne du ranibizumab dans le corps vitré de patients avec DMLA néovasculaire est de 9 jours environ. On suppose que les concentrations sériques du ranibizumab sont 90 000 fois inférieures à celles mesurées dans le corps vitré.
Troubles de la fonction hépatique
Aucune étude spécifique n'est disponible.
Troubles de la fonction rénale
Aucune étude prospective n'a été réalisée pour examiner la pharmacocinétique de Lucentis chez les patients avec une limitation de la fonction rénale. Dans une analyse de pharmacocinétique de population réalisée chez les patients avec DMLA néovasculaire, 68% des patients (n = 136/200) présentaient une limitation de la fonction rénale (pour 46,5% légère, pour 20% modérée et pour 1,5% sévère). Parmi les patients avec OVR examinés, 48,2% (n = 253/525) présentaient une insuffisance rénale (légère dans 36,4% des cas, modérée dans 9,5% des cas et sévère dans 2,3% des cas). La baisse de la clairance systémique du ranibizumab observée était statistiquement non significative.
Population pédiatrique (prématurés atteints de RDP)
Après administration intravitréenne de ranibizumab 0,1 mg dans chaque œil, la concentration sérique de ranibizumab chez les prématurés atteints de RDP était plus élevée que chez les patients adultes atteints de DMLA néovasculaire et traités avec 0,5 mg dans chaque œil. Selon une analyse pharmacocinétique de la population, les différences de Cmax et d'ASCinf étaient environ respectivement 8 et 5 fois supérieures. La demi-vie systémique apparente était d'environ 6 jours. Lors de cette analyse, aucune relation n'a été observée entre la concentration systémique de ranibizumab et celle du VEGF.
Données précliniques
L'administration intravitréenne bilatérale de ranibizumab chez le singe Cynomolgus à des doses variant entre 0,25 mg/œil et 2,0 mg/œil toutes les 2 semaines pendant une période allant jusqu'à 26 semaines a entraîné des effets oculaires dose-dépendants.
Dans le chambre antérieure, une augmentation dose-dépendante des «Flare» et des cellules a été observée, avec un maximum à 2 jours après l'injection. L'intensité de la réaction inflammatoire a généralement régressé avec les injections suivantes ou pendant la phase de récupération, sans être entièrement réversible dans tous les cas. Dans la chambre postérieure, des hémorragies rétiniennes périveineuses focales réversibles sont survenues principalement après la première administration, ainsi que des infiltrations cellulaires vitréennes et des «mouches volantes». Ces dernières ont eu tendance à être dose-dépendantes et ont persisté jusqu'à la fin du traitement. Dans l'étude sur 26 semaines, l'intensité de l'inflammation a augmenté avec le nombre d'injections et le test a dû être arrêté de façon prématurée. Une réversibilité des résultats a été observée pendant la phase de récupération. La formation d'une cataracte a été observée chez certains animaux après une phase relativement longue de forte inflammation, ce qui indique que les modifications du cristallin étaient plutôt secondaires à l'inflammation sévère. Une augmentation transitoire de la pression intraoculaire a été observée après l'injection intravitréenne, indépendamment de la dose.
Les modifications microscopiques au niveau des tissus oculaires ont toutes été liées à l'inflammation et n'ont pas suggéré l'existence de processus dégénératifs dans de quelconques structures oculaires. Dans certains cas, des modifications inflammatoires granulomateuses ont été observées au niveau de la papille du nerf optique. Ces modifications du segment postérieur ont régressé pendant la phase de repos et ont complètement disparu dans certains cas.
Aucun signe de toxicité systémique n'a été observé après une injection intravitréenne induisant des taux sériques plus de 100 fois supérieurs à ceux obtenus lors d'une utilisation thérapeutique normale chez l'être humain. Des anticorps contre le ranibizumab dans le sérum ou dans le corps vitré n'ont pas été observés chez tous les animaux traités.
La documentation préclinique couvre uniquement l'exposition systémique après exposition intravitréenne. On ne dispose d'aucune donnée sur la mutagénicité, la carcinogénicité et l'immunotoxicité de Lucentis chez l'animal.
Toxicité sur la reproduction
Lors d'un traitement intravitréen de singes femelles gestantes avec des doses de ranibizumab correspondant à l'exposition systémique maximale, soit 0,9-7 fois l'exposition clinique la plus élevée imaginable, aucun effet embryo-fœtotoxique ou tératogène et aucun effet sur le poids et la structure du placenta n'ont été relevés; le ranibizumab devra cependant malgré tout être considéré comme potentiellement tératogène et embryo-/ fœtotoxique, compte tenu de ses propriétés pharmacologiques.
L'absence d'effets imputés au ranibizumab sur le développement embryo-fœtal est probablement surtout due au fait que le fragment Fab ne passe pas la barrière placentaire. Un cas de présence de ranibizumab dans le sérum d'un fœtus dont la mère avait des taux sériques élevés de cette substance a néanmoins été rapporté; cela suggère que l'anticorps anti-ranibizumab a servi de protéine de transport (avec région Fc) du ranibizumab, diminuant ainsi la clairance sérique chez la mère et permettant le passage à travers la barrière placentaire. Comme les examens sur le développement embryo-fœtal ont été réalisés chez des animaux gestants sains et que la perméabilité du placenta au fragment Fab pourrait être modifiée par une maladie, l'interprétation de ces résultats impose une certaine prudence.
Remarques particulières
Incompatibilités
Non pertinent.
Aucune étude de tolérance n'ayant été effectuée, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver le médicament hors de portée des enfants.
Flacon: conserver dans l'emballage d'origine, pour protéger le contenu de la lumière. Conserver au réfrigérateur (2–8 °C). Ne pas congeler.
Le flacon non ouvert peut être conservé pendant une durée allant jusqu'à 24 heures à température ambiante (25 °C).
Seringue prête à l'emploi: conserver dans l'emballage blister scellé et dans l'emballage d'origine, pour protéger le contenu de la lumière. Conserver au réfrigérateur (2–8 °C). Ne pas congeler.
Remarques concernant la manipulation
Pour des raisons microbiologiques, la solution injectable prête à l'emploi provenant d'un flacon ou d'une seringue prête à l'emploi doit être utilisée immédiatement après l'ouverture du récipient. La solution non utilisée doit être éliminée (voir «Posologie/Mode d'emploi»). Un flacon ou une seringue prête à l'emploi est destiné à l'administration d'une dose unique.
Flacon (adultes et prématurés)
Le flacon est stérile. Il ne doit pas être utilisé si l'emballage est endommagé. La stérilité du flacon n'est garantie que si l'emballage et son scellage sont intacts. Il ne doit pas être utilisé si la solution est colorée ou trouble ou si elle contient des particules.
Pour la préparation et la réalisation de l'injection intravitréenne, les éléments à usage unique suivants sont nécessaires:
- aiguille avec filtre de 5 µm (18 G)
- seringue stérile de 1 ml
- aiguille à injection (13 mm, 30 Gauge)
Ces éléments ne sont pas inclus dans l'emballage qui ne contient que le flacon. Le paquet qui contient le flacon et l'aiguille avec filtre ne contient ni la seringue stérile de 1 ml, ni l'aiguille à injection.
Seringue prête à l'emploi: la seringue prête à l'emploi Lucentis non ouverte peut, avant utilisation, être entreposée jusqu'à 24 heures à température ambiante (25 °C).
Seringue prête à l'emploi (adultes uniquement)
La seringue prête à l'emploi est uniquement destinée à l'usage unique (voir rubrique 4 «Posologie/Mode d'emploi»).
La seringue prête à l'emploi est stérile. N'utilisez pas la seringue prête à l'emploi si l'emballage est endommagé. La stérilité de la seringue prête à l'emploi n'est garantie que si l'emballage blister est scellé et intact. N'utilisez pas la seringue prête à l'emploi si la solution est colorée ou trouble ou si elle contient des particules.
Veuillez suivre les instructions d'utilisation pour préparer Lucentis en vue d'une administration intravitréenne:
Titre | Instructions | Schéma/Figure |
|---|---|---|
Lisez attentivement toutes les instructions avant d'utiliser la seringue prête à l'emploi.La seringue prête à l'emploi est destinée à l'usage unique. La seringue prête à l'emploi est stérile. Ne pas utiliser un produit dont l'emballage est endommagé. Remarque: la dose doit être ajustée à 0,05 ml. | ||
| Description de la seringue prête à l'emploi |  | |
| Préparation | 1. Assurez-vous que l'emballage contient l'élément suivant:
| (aucun schéma/aucune figure) |
| Contrôle de la seringue | 3. Vérifiez les points suivants:
| |
| Retrait du capuchon de la seringue | 5. Retirez le capuchon de la seringue en l'inclinant sur le côté (ne pas tourner ou visser, voir figure 2). 6. Jetez le capuchon de la seringue (voir figure 3). |   |
| Mise en place de l'aiguille | 7. Mettez en place une aiguille à injection stérile longue d'environ 1,3 cm et de taille 30 G sur la seringue en la vissant fermement sur l'extrémité Luer Lock (voir figure 4). 8. Retirez délicatement le capuchon protecteur de l'aiguille en le tirant verticalement (voir figure 5). Remarque: n'essuyez l'aiguille en aucun cas. | 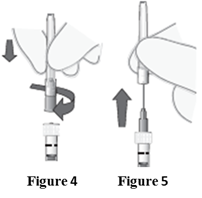 |
| Élimination des bulles d'air | 9. Tenez la seringue en position verticale. 10. Si des bulles d'air sont présentes, tapotez délicatement la seringue avec le doigt jusqu'à ce que les bulles d'air remontent (voir figure 6). |  |
| Ajustement de la dose | 11. Placez la seringue à hauteur des yeux et poussez délicatement le piston, jusqu'à aligner le bord situé juste en-dessous de la partie bombée de la butée en caoutchouc avec le trait indicateur de la dose (voir figure 7).
| 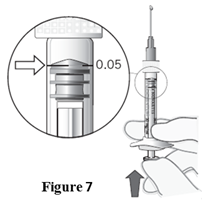 |
| Injection | L'injection doit être réalisée dans des conditions d'asepsie. 12. L'aiguille à injection doit être introduite 3,5‑4,0 mm en arrière du limbe dans le corps vitré en évitant le méridien horizontal et en visant le milieu du globe oculaire. 13. Injectez lentement la solution jusqu'à ce que la butée de caoutchouc touche le bas de la seringue afin d'administrer un volume de 0,05 ml. 14. Pour les injections suivantes, utilisez un autre site d'injection sclérotique. 15. Après l'injection, ne couvrez pas l'aiguille avec le capuchon de sécurité et ne séparez pas l'aiguille de la seringue prête à l'emploi. Jetez la seringue prête à l'emploi utilisée avec l'aiguille dans une boîte à déchets adaptée ou conformément à la réglementation locale. | |
Numéro d’autorisation
57664, 63277 (Swissmedic).
Titulaire de l’autorisation
Novartis Pharma Schweiz AG, Risch; Domicile: 6343 Rotkreuz.
Mise à jour de l’information
Octobre 2020.
Reviews (0)
Recently Viewed


Entecavir Sandoz Filmtabletten 1mg 30 Stück
Was ist Entecavir Sandoz und wann wird es angewendet?Entecavir Sandoz enthält den Wirkstoff Entecavi..
806.40 CHF






Free consultation with an experienced pharmacist
Describe the symptoms or the right drug - we will help you choose its dosage or analogue, place an order with home delivery or just consult.
We are 14 pharmacists and 0 bots. We will always be in touch with you and will be able to communicate at any time.
Bestsellers

Milupa Aptamil Pepti Syneo Can 400g
Product code: 7748334Aptamil Pepti Syneo is a special food for babies from birth who are allergic to cow's milk protein a..
55.08 CHF


Weleda Baby Calendula Wind- und Wetterbalsam 30ml
Product code: 5455461Der Weleda Baby Calendula Wind- und Wetterbalsam schützt die sensible Babyhaut intensiv bei Kälte un..
15.35 CHF


HerpoTherm herpes pen
Product code: 7798882Herpotherm® - the heating pen Cold sores are not only unattractive but can also be very painful. Th..
78.48 CHF



