




Relvar Ellipta Inh PLV 92mcg / 22mcg 30 Dos
Relvar Ellipta Inh Plv 92mcg/22mcg 30 Dos
-
99.50 CHF
- Price in reward points: 3131

- Availability: In stock
- Brand: GLAXO SMITHKLINE AG
- Product Code: 5889797
- ATC-code R03AK10
- EAN 7680629690016
Ingredients:
Magnesium stearat, Lactose-1-Wasser, Fluticason, Fluticason furoat 92 mcg , Vilanterol 22 mcg , Vilanterol trifenatat.

Variants
Description
 Deutsch
Deutsch French
French Italian
ItalianWas ist Relvar Ellipta und wann wird es angewendet?
Relvar Ellipta enthält zwei Arzneimittel – Fluticasonfuroat und Vilanterol – zur Behandlung von Asthma bei Patienten ab 12 Jahren und chronisch-obstruktiver Lungenkrankheit (COPD) bei Erwachsenen ab 40 Jahren. Relvar Ellipta wird mithilfe des Ellipta-Inhalators durch den Mund in die Lungen eingeatmet.
Fluticasonfuroat gehört zur Arzneimittelgruppe der Kortikosteroide, häufig einfach als Steroide bezeichnet. Kortikosteroide wirken entzündungshemmend. Sie bewirken einen Rückgang der Schwellung und der Reizungserscheinungen in den kleinen Luftwegen der Lungen und lindern auf diese Weise die Atembeschwerden. Kortikosteroide tragen ausserdem zur Vorbeugung von Asthmaanfällen und COPD-Exazerbationen bei.
Vilanterol gehört zur Arzneimittelgruppe der Bronchodilatatoren. Es wirkt entspannend auf die Muskulatur der kleinen Luftwege in den Lungen. Dadurch erweitern sich die Luftwege und die Luft kann leichter in die Lunge ein- und wieder ausströmen. Bei regelmässiger Anwendung sorgt das Arzneimittel dafür, dass die kleinen Luftwege durchgängig bleiben.
In Kombination tragen diese beiden Arzneimittel bei regelmässiger Anwendung zur Kontrolle Ihrer Atembeschwerden bei.
Asthma ist eine chronische Lungenerkrankung, bei welcher sich die Muskeln um die kleineren Atemwege zusammenziehen (Bronchokonstriktion) und es zu entzündlichen Schleimhautveränderungen (Schwellung und Reizung) in den Atemwegen kommt. Die Symptome treten episodenhaft auf und umfassen unter anderem Kurzatmigkeit, Giemen (pfeifende Atemgeräusche), Engegefühl in der Brust und Husten.
Bei der chronisch-obstruktiven Lungenkrankheit (COPD) entzündet und verdickt sich die Schleimhaut der Luftwege, in der Regel als Folge des Rauchens. COPD ist eine schleichende Erkrankung mit allmählich fortschreitender Verschlechterung. Zu den Symptomen gehören Kurzatmigkeit, Husten, Beschwerden im Brustkorb und Abhusten von Schleim.
Relvar Ellipta dient zur Langzeitbehandlung und darf nur auf Verschreibung des Arztes bzw. der Ärztin hin angewendet werden.
Wann darf Relvar Ellipta nicht angewendet werden?
Bei Überempfindlichkeit gegenüber einem Bestandteil von Relvar Ellipta (Fluticasonfuroat, Vilanterol, Laktose, Milchproteine, Magnesiumstearat) darf Relvar Ellipta nicht angewendet werden.
Wann ist bei der Anwendung von Relvar Ellipta Vorsicht geboten?
Relvar Ellipta ist nicht zur Behandlung eines akuten Asthmaanfalles geeignet.
Wichtig bei der Behandlung von Atemnot, die plötzlich auftritt oder sich rasch verschlimmert: Wenn zusätzliche Inhalationen Ihres sofortwirkenden Notfall-Inhalators, der Ihnen zusätzlich vom Arzt bzw. von der Ärztin verschrieben worden ist, die Atemnot nicht eindeutig bessern, müssen Sie sofort den Arzt bzw. die Ärztin oder das nächste Spital aufsuchen.
Wenn Sie trotz Behandlung häufiger unter Atemnot oder pfeifenden Atemgeräuschen leiden, so sollten Sie dies Ihrem Arzt bzw. Ihrer Ärztin mitteilen, damit nötigenfalls zusätzliche Massnahmen eingeleitet werden können.
Informieren Sie sofort Ihren Arzt bzw. Ihre Ärztin, wenn Sie während der Behandlung mit Relvar Ellipta verschwommenes Sehen oder andere Augenbeschwerden feststellen, oder wenn Sie unter vermehrtem Durst, häufigem Wasserlassen oder unerklärlicher Müdigkeit leiden (Symptome von erhöhtem Blutzucker).
Bei übermässigem Gebrauch oder Langzeitanwendung bei Jugendlichen kann die Möglichkeit einer Wachstumsverzögerung nicht ausgeschlossen werden. Der Arzt bzw. die Ärztin wird deshalb die Wachstumsentwicklung von Jugendlichen, welche über längere Zeit mit Relvar Ellipta behandelt werden, sorgfältig beobachten.
Wenn Sie gleichzeitig andere Arzneimittel einnehmen, können sich deren Wirkungen gegenseitig beeinflussen, oder es kann zu einem gehäuften Auftreten unerwünschter Wirkungen führen. Die gleichzeitige Einnahme folgender Arzneimittel sollte vermieden werden: Arzneimittel zur Behandlung von gewissen Herzkrankheiten, Depressionen, Allergien, Pilzinfektionen sowie Arzneimittel, die das Immunsystem unterdrücken und bestimmte HIV-Arzneimittel (z.B. Ritonavir, Cobicistat). Nehmen Sie vor der Einnhame eines solchen Arzniemittels Rücksprache mit Ihrem Arzt resp. Ärztin. Asthma- oder COPD-Medikamente, welche einen sogenannten langwirksamen β2-Stimulator enthalten, sollten nicht zusammen mit Relvar Ellipta angewendet werden.
Der Einfluss von Relvar Ellipta auf die Fähigkeit, Fahrzeuge zu führen oder Maschinen zu bedienen, wurde nicht untersucht.
Informieren Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin, wenn Sie
- an anderen Krankheiten leiden (Herzprobleme, erhöhter Blutdruck, Lebererkrankungen, Diabetes, Tuberkulose, chronische oder unbehandelte Infektionen oder einer Lungenentzündung)
- Allergien haben oder
- andere Arzneimittel (auch selbstgekaufte!) einnehmen oder äusserlich anwenden.
Darf Relvar Ellipta während einer Schwangerschaft oder in der Stillzeit angewendet werden?
Wenn Sie schwanger sind oder es werden möchten, dürfen Sie Relvar Ellipta nur anwenden, wenn es Ihr Arzt resp. Ihre Ärztin ausdrücklich erlaubt. Wenn Sie Ihr Kind stillen, dürfen Sie Relvar Ellipta nicht anwenden.
Wie verwenden Sie Relvar Ellipta?
Wenden Sie dieses Arzneimittel immer genau wie mit Ihrem Arzt resp. Ärztin abgesprochen an. Fragen Sie bei Ihrem Arzt resp. Ärztin oder Apotheker resp. Apothekerin nach, wenn Sie sich nicht sicher sind.
Relvar Ellipta sollte täglich, immer zur gleichen Tageszeit inhaliert werden, da es 24 Stunden wirksam ist. Es ist sehr wichtig, dass Sie Relvar Ellipta gemäss den ärztlichen Anweisungen täglich anwenden. Dies trägt dazu bei, dass Sie tagsüber und nachts symptomfrei bleiben.
Wenn Sie merken, dass sich Ihre Atemsymptome nicht bessern, sie häufiger als sonst Atemnot, Husten oder pfeifende Atemgeräusche bekommen, oder wenn Sie Ihren sofortwirkenden Notfall-Inhalator häufiger als sonst benötigen, wenden Sie sich umgehend an Ihren Arzt resp. Ihre Ärztin.
Dosierung bei Asthma:
Erwachsene und Jugendliche ab 12 Jahren: 1-mal täglich 1 Einzeldosis von Relvar Ellipta 92/22.
Bei schwerem Asthma verschreibt Ihnen Ihr Arzt womöglich die stärkere Dosierung von Relvar Ellipta (184/22): 1-mal täglich 1 Einzeldosis von Relvar Ellipta 184/22.
Kinder unter 12 Jahren: Die Sicherheit und Wirksamkeit von Relvar Ellipta bei Kindern unter 12 Jahren ist nicht untersucht und sollte von diesen nicht angewendet werden.
Dosierung bei chronisch-obstruktiver Lungenkrankheit (COPD):
Erwachsene ab 40 Jahren: 1-mal täglich 1 Einzeldosis von Relvar Ellipta 92/22.
Ändern Sie nicht von sich aus die verschriebene Dosierung und hören Sie nicht von sich aus mit der Behandlung auf. Wenn Sie glauben, das Arzneimittel wirke zu schwach oder zu stark, so sprechen Sie mit Ihrem Arzt oder Apotheker bzw. Ihrer Ärztin oder Apothekerin.
Bitte lesen Sie die Gebrauchsanleitung vor der ersten Anwendung genau durch, eine korrekte Anwendung ist sehr wichtig.
Wenn Sie versehentlich eine grössere Menge Relvar Ellipta angewendet haben, als Ihr Arzt resp. Ihre Ärztin Ihnen verordnet hat, wenden Sie sich an Ihren Arzt resp. Ärztin oder Apotheker resp. Apothekerin. Vielleicht merken Sie, dass Ihr Herz schneller schlägt als sonst, dass Sie zittrig sind oder Kopfschmerzen bekommen.
Wenn Sie über längere Zeit grössere Mengen angewendet haben, als Sie sollten, ist es besonders wichtig, dass Sie Ihren Arzt resp. Ärztin oder Apotheker resp. Apothekerin informieren.
Wenden Sie nicht die doppelte Menge an, wenn Sie die vorherige Anwendung vergessen haben. Wenden Sie einfach die nächste Dosis zum üblichen Zeitpunkt an.
Gebrauchsanweisung:
Bitte lesen Sie die Gebrauchsanweisung vor der ersten Anwendung genau durch, eine korrekte Anwendung ist sehr wichtig.
Die Verpackung Ihres Ellipta-Inhalators enthält folgende Teile:
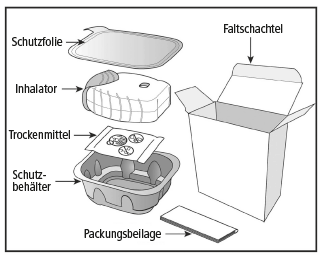
Der Inhalator ist in einem Schutzbehälter verpackt. Bitte öffnen Sie diesen erst, wenn Sie bereit sind für die Inhalation. Dann entfernen Sie die Schutzfolie, um den Behälter zu öffnen. Dieser enthält ausserdem ein Trockenmittelpäckchen zur Feuchtigkeitsreduktion. Bitte entsorgen Sie das Trockenmittelpäckchen. Dieses darf nicht geöffnet, geschluckt oder eingeatmet werden.
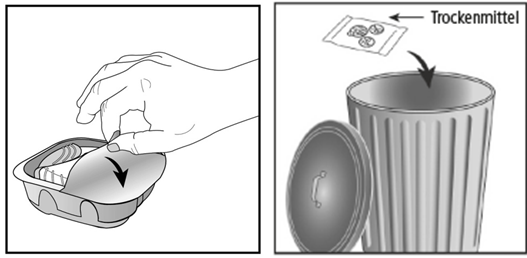
Bei der Entnahme aus der Verpackung befindet sich der Inhalator in geschlossener Position. Öffnen Sie den Inhalator nur zur Inhalation einer Dosis und erst, wenn Sie bereit sind für die Inhalation.
Schreiben Sie das Öffnungsdatum des Schutzbehälters auf die Etikette des Inhalators, sobald Sie diesen aus dem Schutzbehälter genommen haben. Nach dem Öffnen des Schutzbehälters ist der Inhalator 6 Wochen haltbar. Danach darf der Inhalator nicht mehr verwendet werden.
Bitte lesen Sie vor der ersten Anwendung den folgenden Abschnitt
Wenn Sie die Schutzabdeckung öffnen und schliessen, ohne das Arzneimittel zu inhalieren, ist diese Dosis verloren.
Die Dosis verbleibt zwar im Inhalator, kann jedoch nicht mehr inhaliert werden.
Die versehentliche Verabreichung einer zu hohen oder einer doppelten Dosis in einem Inhalationsvorgang ist dadurch ausgeschlossen.
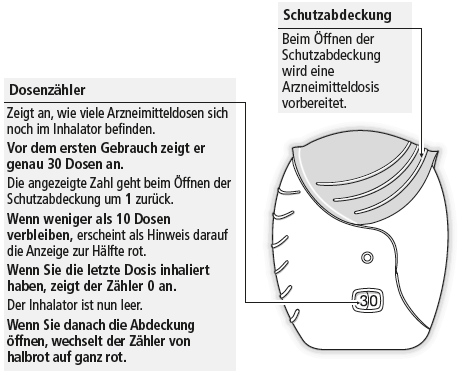
1. Vorbereitung einer Dosis
Warten Sie mit dem Öffnen der Schutzabdeckung, bis Sie zur Inhalation der Dosis bereit sind.
Schieben Sie die Schutzabdeckung ganz nach unten, bis Sie ein Klicken hören.
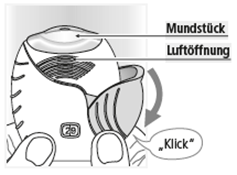
Das Arzneimittel ist nun bereit für die Inhalation.
Die vom Dosenzähler angezeigte Zahl verringert sich dabei um 1.
- Der Inhalator darf auf keinen Fall geschüttelt werden.
- Wenn sich die angezeigte Zahl trotz hörbarem Klicken nicht verringert, gibt der Inhalator kein Arzneimittel ab. Bringen Sie in diesem Fall den Inhalator in die Apotheke zurück und fragen Sie Ihren Apotheker resp. Ihre Apothekerin um Rat.
2. Inhalation des Arzneimittels
Atmen Sie zunächst so tief aus, wie es ohne Anstrengung für Sie möglich ist; der Mund muss dabei vom Inhalator abgewendet sein.
Atmen Sie nicht in den Inhalator aus.
Umschliessen Sie das Mundstück fest mit Ihren Lippen.
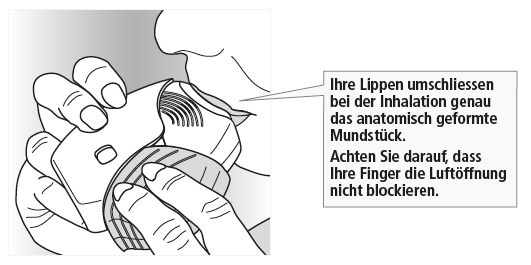
Atmen Sie tief, lange und gleichmässig ein. Halten Sie den Atem anschliessend so lange wie möglich an (mindestens 3 bis 4 Sekunden).
Entfernen Sie den Inhalator von Ihrem Mund.
Atmen Sie langsam und vorsichtig aus.
Auch bei korrekter Anwendung des Inhalators kann es sein, dass Sie das Arzneimittel nicht schmecken bzw. spüren.
3. Schliessen des Inhalators und Ausspülen des Mundes
Soll das Mundstück vor dem Schliessen der Schutzabdeckung gereinigt werden, verwenden Sie dazu ein trockenes Tuch.
Die Schutzabdeckung so weit wie möglich nach oben schieben, bis das Mundstück wieder vollständig abgedeckt ist.
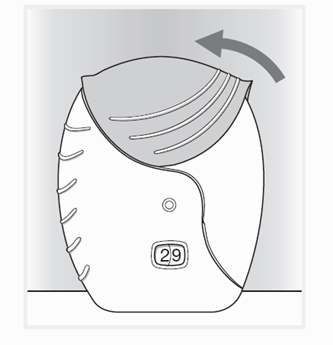
Spülen Sie nach Gebrauch des Inhalators Ihren Mund mit Wasser aus. Das verringert die Wahrscheinlichkeit von Nebenwirkungen wie wunden Stellen in Mund und Rachen.
Hinweis:
- Wenn das Anzeigefeld des Dosenzählers zur Hälfte rot ist, wird es Zeit, dass Sie sich einen neuen Inhalator besorgen. Wenn das Anzeigefeld des Dosenzählers komplett rot ist, ist der Inhalator leer und Sie benötigen einen neuen.
- Versuchen Sie nicht, den Dosenzähler zu manipulieren oder vom Inhalatorgehäuse zu entfernen. Der Zähler kann nicht zurückgesetzt und nicht vom Inhalator abgenommen werden.
- Wenn Ihr Arzt resp. Ihre Ärztin Ihnen andere Anweisungen zur Anwendung Ihres Inhalators gegeben hat, so halten Sie sich bitte genau daran. Falls Sie Probleme mit der Anwendung haben, wenden Sie sich bitte an Ihren Arzt resp. Ärztin oder Apotheker bzw. Apothekerin.
Welche Nebenwirkungen kann Relvar Ellipta haben?
Plötzliche Atmungsschwierigkeiten nach dem Gebrauch von Relvar Ellipta sind selten (betrifft weniger als 1 Person/1000). Wenn Ihre Atemnot oder die pfeifenden Atemgeräusche sich unmittelbar nach der Anwendung von Relvar Ellipta verschlechtern, setzen Sie das Arzneimittel bitte sofort ab und wenden Sie sich möglichst unverzüglich an Ihren Arzt resp. Ihre Ärztin.
Folgende Nebenwirkungen können bei der Anwendung von Relvar Ellipta zusätzlich auftreten:
Sehr häufig (bei mehr als 1 von 10 Patienten) kann es während der Behandlung zu Kopfschmerzen oder grippeähnlichen Symptomen kommen.
Häufig (bei bis zu 1 von 10 Patienten) kann es zu Pilzinfektionen im Mund und Rachen kommen. Dieser Nebenwirkung können Sie entgegenwirken, indem Sie Ihren Mund nach der Anwendung von Relvar Ellipta umgehend mit Wasser ausspülen.
Ebenfalls häufig wurde über Entzündungen der unteren Atemwege (Bronchitis), Infektionen und Entzündungen der Nasennebenhöhlen oder des Rachens, Grippe, Schmerzen und Reizungserscheinungen im hinteren Bereich der Mundhöhle und im Rachen, juckende, laufende oder verstopfte Nase, Husten, Heiserkeit sowie über Lungenentzündung berichtet. Lungenentzündungen sind häufiger bei COPD-Patienten als bei Asthma-Patienten. Benachrichtigen Sie bitte Ihren Arzt, wenn Sie unter der Behandlung mit Relvar Ellipta eines der folgenden möglichen Symptome einer Lungenentzündung bei sich bemerken: Fieber, Schüttelfrost, verstärkter Auswurf, Änderung der Auswurffarbe, vermehrtes Husten oder verstärkte Atemnot.
Häufig wurde auch über Osteoporose mit Gefahr von Knochenbrüchen, Bauchschmerzen, Rückenschmerzen, Gelenkschmerzen Muskelkrämpfe sowie über Fieber berichtet.
Gelegentlich (bei bis zu 1 von 100 Patienten) kann es zu unregelmässigem oder beschleunigtem Herzschlag, sowie zu Herzklopfen oder erhöhtem Blutzucker kommen.
Selten (bei bis zu 1 von 1000 Patienten) kann es zu psychischen Störungen wie Hyperaktivität, Schlafstörungen, Angst, Depressionen oder Aggressionen kommen.
Selten wurden auch allergische Reaktionen beobachtet. Setzen Sie das Arzneimittel bitte sofort ab und wenden Sie sich möglichst unverzüglich an Ihren Arzt resp. Ihre Ärztin, wenn Sie nach der Anwendung von Relvar eines der folgenden Symptome einer möglichen allergischen Reaktion bei sich bemerken: Hautausschlag oder -rötung, Schwellungen (gelegentlich auch von Gesicht oder Mund, sog. Angioödem), plötzliche Atmungsschwierigkeiten wie Atemnot, pfeifende Atmung oder Husten, plötzliches Schwächegefühl oder Benommenheit (was bis zum Kollaps oder zur Bewusstlosigkeit führen könnte).
Ebenfalls wurde über Angst und Zittern berichtet.
Im Falle einer Überdosierung sollten Sie unverzüglich einen Arzt bzw. eine Ärztin konsultieren.
Wenn Sie Nebenwirkungen bemerken, die hier nicht beschrieben sind, sollten Sie Ihren Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin informieren.
Was ist ferner zu beachten?
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Relvar Ellipta ausserhalb der Reichweite von Kindern, nicht über 25 °C und in der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen. Entfernen Sie den Foliendeckel erst unmittelbar vor dem ersten Gebrauch des Inhalators.
Haltbarkeit nach Öffnen des Schutzbehälters: 6 Wochen.
Das Datum, an dem der Schutzbehälter geöffnet worden ist, sollten Sie auf der Etikette des Inhalators vermerken, sobald Sie diesen aus dem Schutzbehälter genommen haben.
Weitere Auskünfte erteilt Ihnen Ihr Arzt oder Apotheker bzw. Ihre Ärztin oder Apothekerin. Diese Personen verfügen über die ausführliche Fachinformation.
Was ist in Relvar Ellipta enthalten?
Relvar Ellipta 92/22 enthält als Wirkstoffe Fluticasonfuroat (92 µg pro abgegebener Dosis) und Vilanterol (22 µg pro abgegebener Dosis) sowie als Hilfsstoffe Laktose (mit geringen Mengen an Milchproteinen) und Magnesiumstearat.
Relvar Ellipta 184/22 enthält als Wirkstoffe Fluticasonfuroat (184 µg pro abgegebener Dosis) und Vilanterol (22 µg pro abgegebener Dosis) sowie als Hilfsstoffe Laktose (mit geringen Mengen an Milchproteinen) und Magnesiumstearat.
Zulassungsnummer
62969 (Swissmedic).
Wo erhalten Sie Relvar Ellipta? Welche Packungen sind erhältlich?
Relvar Ellipta ist in Apotheken nur auf ärztliche Verschreibung erhältlich.
Relvar Ellipta 92/22
Packung mit 1 Ellipta Inhalator zu 30 Einzeldosen
Packung mit 3 Ellipta Inhalatoren zu 30 Einzeldosen
Relvar Ellipta 184/22
Packung mit 1 Ellipta Inhalator zu 30 Einzeldosen
Packung mit 3 Ellipta Inhalatoren zu 30 Einzeldosen
Zulassungsinhaberin
GlaxoSmithKline AG, 3053 Münchenbuchsee.
Diese Packungsbeilage wurde im Januar 2019 letztmals durch die Arzneimittelbehörde (Swissmedic) geprüft.
Qu’est-ce que Relvar Ellipta et quand doit-il être utilisé?
Relvar Ellipta contient deux principes actifs, le furoate de fluticasone et le vilantérol, pour le traitement de l'asthme chez les patients à partir de 12 ans et le traitement de la bronchopneumopathie chronique obstructive (BPCO) chez l'adulte à partir de 40 ans. Relvar Ellipta est inhalé dans les poumons par la bouche à l'aide de l'inhalateur Ellipta.
Le furoate de fluticasone fait partie du groupe de médicaments appelés corticostéroïdes, voire souvent simplement corticoïdes. Les corticostéroïdes ont des effets anti-inflammatoires. Ils provoquent une régression des gonflements et des manifestations d'irritation dans les petites ramifications des voies respiratoires pulmonaires et soulagent ainsi les symptômes respiratoires. Les corticostéroïdes contribuent aussi à prévenir les crises d'asthme et les exacerbations de la BPCO.
Le vilantérol fait partie du groupe des médicaments bronchodilatateurs. Il provoque une relaxation de la musculature entourant les petites voies respiratoires pulmonaires. Cela permet une dilatation des voies respiratoires et facilite l'entrée et la sortie d'air dans les poumons. Utilisé régulièrement, le médicament veille à maintenir les petites voies respiratoires suffisamment ouvertes.
L'utilisation régulière de ces deux agents actifs associés contribue au contrôle de vos troubles respiratoires.
L'asthme est une maladie pulmonaire chronique dans laquelle la musculature entourant les petites voies respiratoires se contracte (bronchoconstriction) et la muqueuse tapissant les voies aériennes est modifiée par des processus inflammatoires (gonflement et irritation). Les symptômes – incluant entre autres un souffle court, des sibilances (sifflements respiratoires), une sensation d'oppression thoracique et de la toux – se manifestent de façon épisodique.
Dans la bronchopneumopathie chronique obstructive (BPCO), généralement due au tabagisme, la muqueuse respiratoire est enflammée et épaissie. La BPCO est une maladie insidieuse qui se développe et s'aggrave lentement de façon progressive. Ses symptômes englobent un souffle court, de la toux, des symptômes thoraciques et l'expectoration de glaires.
Relvar Ellipta est utilisé pour le traitement au long cours, uniquement sur prescription du médecin.
Quand Relvar Ellipta ne doit-il pas être utilisé?
Relvar Ellipta ne doit pas être utilisé en cas d'hypersensibilité à l'un des composants de Relvar Ellipta (furoate de fluticasone, vilantérol, lactose, protéines du lait, stéarate de magnésium).
Quelles sont les précautions à observer lors de l’utilisation de Relvar Ellipta?
Relvar Ellipta ne convient pas au traitement de la crise d'asthme aiguë.
Conseil important pour le traitement d'une détresse respiratoire se manifestant soudainement ou s'aggravant rapidement: si des inhalations complémentaires de votre inhalateur de secours à action immédiate, prescrit en supplément par votre médecin, ne conduisent pas à un soulagement efficace de la détresse respiratoire, vous devez immédiatement vous rendre chez votre médecin ou à l'hôpital le plus proche.
Si vous souffrez plus fréquemment de détresse respiratoire ou de sifflements respiratoires malgré le traitement, vous devez prévenir votre médecin pour qu'il puisse prendre des mesures complémentaires au besoin.
Informez immédiatement votre médecin si, pendant votre traitement par Relvar Ellipta, vous développez une vision floue ou d'autres troubles oculaires, ou si vous présentez une soif accrue, un besoin plus fréquent d'uriner ou une fatigue inexpliquée (symptômes d'une glycémie élevée).
Dans le cas d'une utilisation excessive ou à long terme chez l'adolescent, l'éventualité d'un retard de croissance ne peut pas être exclue. C'est pourquoi le médecin observera attentivement l'évolution de la croissance chez les adolescents traités par Relvar Ellipta pendant une période prolongée.
Si vous prenez en même temps d'autres médicaments, il est possible que les différents médicaments s'influencent mutuellement ou que des effets indésirables surviennent plus fréquemment. Vous devez éviter de prendre en même temps les médicaments suivants: médicaments utilisés pour traiter certaines maladies cardiaques, la dépression, les allergies ou les mycoses ainsi que les médicaments qui affaiblissent le système immunitaire ou certains médicaments anti-VIH (p.ex. ritonavir, cobicistat). Contactez votre médecin avant de prendre un tel médicament. Certains médicaments contre l'asthme ou la BPCO contenant ce que l'on appelle un β2-stimulant de longue durée d'action, ne doivent pas être utilisés en même temps que Relvar Ellipta.
L'influence de Relvar Ellipta sur l'aptitude à conduire et la capacité à utiliser des machines n'a pas été étudiée.
Veuillez informer votre médecin ou votre pharmacien si
- vous souffrez d'une autre maladie (problèmes cardiaques, hypertension artérielle, maladie du foie, diabète, tuberculose, infections chroniques ou non traitées ou pneumonie),
- vous êtes allergique ou
- vous prenez ou utilisez déjà d'autres médicaments en usage externe (même en automédication!)
Relvar Ellipta peut-il être utilisé pendant la grossesse ou l’allaitement?
Si vous êtes enceinte ou envisagez une grossesse, vous ne devez utiliser Relvar Ellipta que si votre médecin vous y autorise expressément. Vous ne devez pas utiliser Relvar Ellipta si vous allaitez.
Comment utiliser Relvar Ellipta?
Utilisez ce médicament toujours exactement selon les instructions de votre médecin. En cas de doutes, demandez des précisions à votre médecin ou à votre pharmacien.
Relvar Ellipta doit être inhalé une fois par jour, toujours à la même heure de la journée, étant donné qu'il agit pendant 24 heures. Il est très important que vous utilisiez Relvar Ellipta quotidiennement, conformément aux instructions du médecin. Cela contribue à éviter la survenue de symptômes au cours de la journée et pendant la nuit.
Veuillez consulter immédiatement votre médecin si vous remarquez que vos symptômes respiratoires ne s'améliorent pas, que vos épisodes de détresse respiratoire, de toux ou de sifflements respiratoires deviennent plus fréquents ou que vous avez besoin plus souvent qu'à l'ordinaire de votre inhalateur de secours à action rapide.
Posologie pour le traitement de l'asthme:
Adultes et adolescents à partir de 12 ans: 1 dose unitaire de Relvar Ellipta 92/22 1 fois par jour.
Dans le cas d'un asthme sévère, votre médecin vous prescrira éventuellement le dosage plus élevé de Relvar Ellipta (184/22): 1 dose unitaire de Relvar Ellipta 184/22 1 fois par jour.
Enfants de moins de 12 ans: l'efficacité et la sécurité de Relvar Ellipta n'ont pas été étudiées chez les enfants de moins de 12 ans, ceux-ci ne doivent donc pas l'utiliser.
Posologie pour le traitement de la bronchopneumopathie chronique obstructive (BPCO):
Adultes à partir de 40 ans: 1 dose unitaire de Relvar Ellipta 92/22 1 fois par jour.
Ne changez pas le dosage prescrit et n'arrêtez pas le traitement de votre propre chef. Adressez-vous à votre médecin ou à votre pharmacien si vous estimez que l'efficacité du médicament est trop faible ou au contraire trop forte.
Lisez soigneusement le mode d'emploi avant la première utilisation, car l'utilisation correcte joue un rôle très important.
Si vous avez utilisé accidentellement une plus grande quantité de Relvar Ellipta que celle prescrite par votre médecin, veuillez contacter votre médecin ou votre pharmacien. Vous remarquerez éventuellement que votre cœur bat plus rapidement que d'habitude, que vous tremblez ou que vous avez mal à la tête.
Si vous avez utilisé des quantités trop élevées pendant une période prolongée, il est particulièrement important d'informer votre médecin ou votre pharmacien.
N'utilisez pas une double dose si vous avez oublié la dose précédente. Inhalez simplement la prochaine dose à l'heure habituelle.
Mode d'emploi:
Lisez attentivement le mode d'emploi avant la première utilisation, car l'utilisation correcte joue un rôle très important.
L'emballage de votre inhalateur Ellipta contient les éléments suivants:
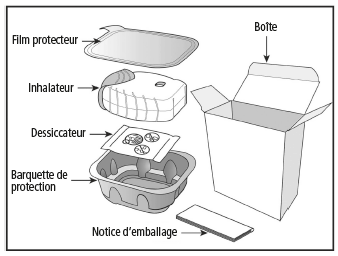
L'inhalateur est emballé dans une barquette de protection. N'ouvrez celle-ci que lorsque vous êtes prêt(e) à faire l'inhalation. Retirez ensuite le film protecteur pour ouvrir la barquette. Celle-ci contient en outre un élément dessiccateur pour réduire l'humidité. Jetez le dessiccateur. Celui-ci ne doit pas être ouvert, avalé ou aspiré.
Lorsque vous retirez l'inhalateur de son emballage, il est en position fermée. N'ouvrez l'inhalateur que pour l'inhalation d'une dose et seulement lorsque vous êtes prêt(e) faire à l'inhalation.
Inscrivez sur l'étiquette de l'inhalateur la date d'ouverture de la barquette de protection dès que vous avez retiré l'inhalateur de la barquette de protection. L'inhalateur se conserve 6 semaines après l'ouverture de la barquette de protection. Ensuite l'inhalateur ne doit plus être utilisé.
Lisez attentivement la section suivante avant la première utilisation
Si vous ouvrez et refermez le couvercle protecteur sans inhaler le médicament, la dose chargée sera perdue.
Cette dose restera dans l'inhalateur, mais ne pourra plus être inhalée.
Ainsi, l'administration accidentelle d'une dose trop élevée ou d'une double dose en une seule inhalation est exclue.
1. Préparation d'une dose
N'ouvrez pas le couvercle protecteur avant d'être prêt(e) à l'inhalation de la dose.
Poussez à fond le couvercle protecteur vers le bas jusqu'à ce que l'enclenchement soit confirmé par un «clic» audible.
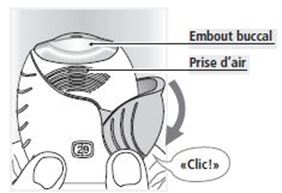
Le médicament être maintenant prêt pour l'inhalation.
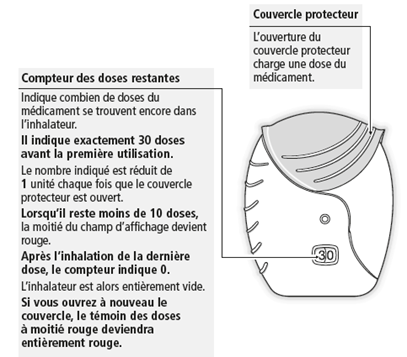
Le nombre indiqué par le compteur des doses restantes est alors réduit de 1 unité.
- L'inhalateur ne doit en aucun cas être secoué.
- Si le nombre indiqué par le compteur des doses restantes ne baisse pas malgré le «clic» audible, l'inhalateur ne délivrera pas le médicament. Dans un tel cas, vous devez rapporter l'inhalateur à la pharmacie et demander conseil à votre pharmacien.
2. Inhalation du médicament
Expirez aussi profondément que vous le pouvez sans forcer, pendant l'expiration la bouche doit être détournée de l'inhalateur.
N'expirez pas dans l'inhalateur.
Entourez fermement l'embout buccal entre vos lèvres.

Inspirez profondément, longtemps et régulièrement. Retenez ensuite votre souffle aussi longtemps que possible (au moins 3 à 4 secondes).
Retirez l'inhalateur de votre bouche.
Expirez doucement et lentement.
Même dans le cas d'une utilisation correcte de l'inhalateur, il est possible que vous ne ressentiez pas l'inhalation du médicament ou le goût du médicament.
3. Fermeture de l'inhalateur et rinçage de votre bouche
Si vous souhaitez nettoyer l'embout buccal avant de fermer le couvercle protecteur, faites-le avec un linge sec.
Poussez le couvercle protecteur vers le haut jusqu'au bout, de sorte à couvrir entièrement l'embout buccal.
Après l'inhalation, rincez votre bouche à l'eau. Cela réduit le risque d'effets indésirables tels qu'une formation de lésions dans la bouche ou dans la gorge.
Remarque:
- Quand la moitié du champ d'affichage du compteur des doses restantes est rouge, il est temps de vous procurer un nouvel inhalateur. Si le champ d'affichage du compteur des doses restantes est entièrement rouge, l'inhalateur est vide et vous devez en utiliser un nouveau.
- Ne tentez pas de manipuler le compteur des doses restantes ou de l'enlever du boîtier de l'inhalateur. Le compteur ne peut pas être remis à une valeur précédente ou retiré de l'inhalateur.
- Si le médecin vous a donné d'autres instructions pour l'utilisation de l'inhalateur, vous devez les respecter scrupuleusement. En cas de problèmes d'utilisation, veuillez vous adresser à votre médecin ou votre pharmacien.
Quels effets secondaires Relvar Ellipta peut-il provoquer?
Des difficultés respiratoires soudaines sont rares après l'utilisation de Relvar Ellipta (elles touchent moins de 1 personne sur 1000). Si votre détresse respiratoire ou vos sifflements respiratoires s'aggravent immédiatement après l'administration de Relvar Ellipta, cessez immédiatement d'utiliser le médicament et contactez votre médecin le plus rapidement possible.
L'utilisation de Relvar Ellipta peut de plus provoquer les effets indésirables suivants:
Des maux de tête et des symptômes pseudo-grippaux peuvent survenir très fréquemment (chez plus de 1 patient sur 10) au cours du traitement.
On observe fréquemment (chez jusqu'à 1 patient sur 10) des mycoses dans la bouche et dans la gorge. Vous pouvez prévenir cet effet indésirable en vous rinçant la bouche à l'eau immédiatement après l'administration de Relvar Ellipta.
On a également rapporté fréquemment des inflammations des voies respiratoires inférieures (bronchites), des infections et des inflammations des sinus paranasaux ou de la gorge, une grippe, des douleurs et des signes d'irritations dans le fond de la cavité buccale et dans la gorge, un nez qui picote, qui coule ou qui est bouché, de la toux, de l'enrouement, ainsi que des pneumonies. Les pneumonies sont plus fréquentes chez les patients atteints de BPCO que chez les patients asthmatiques. Prévenez votre médecin si vous présentez, au cours de votre traitement par Relvar Ellipta, l'un des éventuels symptômes suivants d'une pneumonie: fièvre, frissons, augmentation des crachats, modification de la couleur des crachats, toux accrue ou aggravation de la détresse respiratoire.
Une ostéoporose avec risque de fractures, des douleurs abdominales, des douleurs dorsales, des douleurs articulaires, des crampes musculaires ainsi que de la fièvre ont aussi été observées fréquemment.
Des battements de cœur irréguliers ou accélérés ainsi que des palpitations ou un taux élevé de sucre dans le sang peuvent survenir occasionnellement (chez jusqu'à 1 patient sur 100).
Dans de rares cas (chez jusqu'à 1 patient sur 1000), des troubles psychiques tels qu'hyperactivité, troubles du sommeil, anxiété, dépression ou agressivité peuvent être observés.
Des réactions allergiques ont également été observées dans de rares cas. Arrêtez immédiatement l'utilisation du médicament et adressez-vous dès que possible à votre médecin si vous présentez l'un des symptômes suivants d'une éventuelle réaction allergique après l'administration de Relvar: éruption ou rougeur cutanée, gonflements (éventuellement aussi du visage ou de la bouche, on parle alors d'un angiœdème), difficultés respiratoires soudaines telles que détresse respiratoire, respiration sifflante ou toux, sensation soudaine de faiblesse ou d'étourdissement (pouvant éventuellement aller jusqu'à un collapsus ou une perte de conscience).
Une anxiété et des tremblements ont également été rapportés.
En cas de surdosage, vous devez consulter un médecin immédiatement.
Si vous remarquez des effets secondaires qui ne sont pas mentionnés dans cette notice, veuillez en informer votre médecin ou votre pharmacien.
À quoi faut-il encore faire attention?
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Conserver Relvar Ellipta hors de portée des enfants. Ne pas conserver au-dessus de 25 °C. Conserver dans l'emballage d'origine pour le protéger de l'humidité. Le film protecteur doit être retiré juste avant la première utilisation de l'inhalateur.
Conservation après ouverture de la barquette de protection: 6 semaines.
Dès que vous avez retiré l'inhalateur de la barquette de protection, vous devez inscrire sur l'étiquette de l'inhalateur la date d'ouverture de la barquette de protection. Pour de plus amples renseignements, consultez votre médecin ou votre pharmacien, qui disposent d'une information détaillée destinée aux professionnels.
Que contient Relvar Ellipta?
Relvar Ellipta 92/22 contient les principes actifs furoate de fluticasone (92 µg par dose délivrée) et vilantérol (22 µg par dose délivrée) ainsi que du lactose (avec de faibles quantités de protéines du lait) et du stéarate de magnésium comme excipients.
Relvar Ellipta 184/22 contient les principes actifs furoate de fluticasone (184 µg par dose délivrée) et vilantérol (22 µg par dose délivrée) ainsi que du lactose (avec de faibles quantités de protéines du lait) et du stéarate de magnésium comme excipients.
Numéro d’autorisation
62969 (Swissmedic).
Où obtenez-vous Relvar Ellipta? Quels sont les emballages à disposition sur le marché?
En pharmacie, sur ordonnance médicale.
Relvar Ellipta 92/22
Emballage de 1 inhalateur Ellipta contenant 30 doses unitaires.
Emballage de 3 inhalateurs Ellipta contenant chacun 30 doses unitaires.
Relvar Ellipta 184/22
Emballage de 1 inhalateur Ellipta contenant 30 doses unitaires.
Emballage de 3 inhalateurs Ellipta contenant chacun 30 doses unitaires.
Titulaire de l’autorisation
GlaxoSmithKline AG, 3053 Münchenbuchsee.
Cette notice d'emballage a été vérifiée pour la dernière fois en janvier 2019 par l'autorité de contrôle des médicaments (Swissmedic).
Che cos’è Relvar Ellipta e quando si usa?
Relvar Ellipta contiene due principi attivi (fluticasone furoato e vilanterolo) per il trattamento dell'asma nei pazienti a partire da 12 anni di età e della broncopneumopatia cronica ostruttiva (BPCO) negli adulti a partire da 40 anni di età. Relvar Ellipta viene inspirato nei polmoni attraverso la bocca, utilizzando l'apposito inalatore Ellipta.
Il fluticasone furoato appartiene al gruppo terapeutico dei corticosteroidi, spesso detti semplicemente steroidi. I corticosteroidi esplicano un'azione antinfiammatoria. Riducono la congestione e i fenomeni irritativi delle piccole vie aeree dei polmoni e pertanto alleviano i disturbi respiratori. Inoltre, i corticosteroidi contribuiscono a prevenire gli attacchi di asma e le esacerbazioni di BPCO.
Il vilanterolo fa parte del gruppo terapeutico dei broncodilatatori. Ha un effetto rilassante sulla muscolatura delle piccole vie aeree dei polmoni. In tal modo le vie aeree si dilatano e l'aria può essere inspirata ed espirata più facilmente dai polmoni. Con un impiego regolare, questo principio attivo mantiene la pervietà delle piccole vie aeree.
L'impiego regolare di questi due principi attivi in combinazione contribuisce a controllare i disturbi respiratori.
L'asma è una malattia polmonare cronica in cui i muscoli intorno alle piccole vie aeree si contraggono (broncocostrizione) e si verificano alterazioni infiammatorie delle mucose delle vie respiratorie (congestione e irritazione). I sintomi si manifestano a episodi e comprendono, fra gli altri, respiro corto, respiro sibilante, senso di costrizione al torace e tosse.
Con la broncopneumopatia cronica ostruttiva (BPCO) la mucosa delle vie respiratorie si infiamma e si ispessisce, generalmente come conseguenza del fumo. La BPCO è una malattia cronica, che peggiora progressivamente. I suoi sintomi comprendono respiro corto, tosse, disturbi al torace ed espettorazione.
Relvar Ellipta è concepito per la terapia a lungo termine e deve essere utilizzato esclusivamente su prescrizione medica.
Quando non si può usare Relvar Ellipta?
Non si deve usare Relvar Ellipta in caso di ipersensibilità a uno dei suoi componenti (fluticasone furoato, vilanterolo, lattosio, lattoproteine, magnesio stearato).
Quando è richiesta prudenza nell’uso di Relvar Ellipta?
Relvar Ellipta non è adatto per il trattamento degli attacchi acuti di asma.
Avvertenza importante in caso di difficoltà respiratorie che si manifestano improvvisamente o si aggravano rapidamente: se le difficoltà respiratorie non migliorano nettamente dopo inalazioni supplementari con l'inalatore di emergenza a effetto immediato prescrittole in aggiunta dal medico, deve recarsi immediatamente dal medico o nel più vicino ospedale.
Se, malgrado il trattamento, soffrisse frequentemente di difficoltà respiratorie o respiro sibilante, dovrebbe comunicarlo al suo medico, in modo che, se necessario, si possano prendere misure supplementari.
Informi subito il suo medico se durante il trattamento con Relvar Ellipta compaiono visione confusa o altri disturbi oculari, oppure se soffre di aumento della sete, urinazione frequente o stanchezza inspiegabile (sintomi di un aumento della glicemia).
In caso di uso eccessivo o prolungato da parte di adolescenti, non si possono escludere ritardi nella crescita. Pertanto il medico seguirà con attenzione lo sviluppo di adolescenti trattati per lunghi periodi con Relvar Ellipta.
Se assume contemporaneamente altri medicamenti, le loro azioni possono influenzarsi reciprocamente, oppure si possono manifestare con maggiore frequenza effetti indesiderati. L'assunzione contemporanea dei medicamenti seguenti deve essere evitata: i medicamenti per il trattamento di determinate malattie cardiache, depressioni, allergie e infezioni micotiche, nonché i medicamenti che sopprimono il sistema immunitario e determinati medicamenti contro l'HIV (per es. ritonavir, cobicistat). Prima di assumere un medicamento di questi consulti il suo medico. I medicamenti per il trattamento dell'asma o della BPCO che contengono un cosiddetto β2-agonista a lunga durata d'azione non devono essere utilizzati in concomitanza con Relvar Ellipta.
L'influsso di Relvar Ellipta sulla capacità di condurre veicoli o usare macchine non è stato oggetto di studi specifici.
Informi il suo medico o il suo farmacista nel caso in cui:
- soffra di altre malattie (problemi cardiaci, ipertensione arteriosa, malattie epatiche, diabete, tubercolosi, infezioni croniche o non trattate o polmonite),
- soffra di allergie o
- assuma o applichi esternamente altri medicamenti (anche se acquistati di sua iniziativa!).
Si può usare Relvar Ellipta durante la gravidanza o l’allattamento?
Se lei è incinta o desidera iniziare una gravidanza, deve usare Relvar Ellipta solo a condizione che sia stato permesso espressamente dal suo medico. Durante l'allattamento non si deve usare Relvar Ellipta.
Come usare Relvar Ellipta?
Usi questo medicamento sempre esattamente come concordato con il suo medico. Se ha qualche dubbio, consulti il suo medico o il suo farmacista.
Relvar Ellipta deve essere inalato tutti i giorni sempre alla stessa ora, dato che il suo effetto ha una durata di 24 ore. È molto importante che usi Relvar Ellipta quotidianamente attenendosi scrupolosamente alle istruzioni del medico. Questo contribuisce a far sì che i sintomi non si manifestino né di giorno né di notte.
Se notasse che i sintomi respiratori non migliorano o che le difficoltà respiratorie, la tosse o il respiro sibilante si manifestano più frequentemente del solito o se dovesse fare ricorso con maggiore frequenza del solito al suo inalatore di emergenza a effetto immediato, consulti immediatamente il suo medico.
Dosaggio in caso di asma:
Adulti e adolescenti a partire da 12 anni di età: 1 dose singola di Relvar Ellipta 92/22 1 volta al giorno.
In caso di asma grave, il medico può eventualmente prescriverle il dosaggio più elevato di Relvar Ellipta (184/22): 1 dose singola di Relvar Ellipta 184/22 1 volta al giorno.
Bambini di età inferiore a 12 anni: la sicurezza e l'efficacia di Relvar Ellipta in caso di bambini di età inferiore ai 12 anni non sono state oggetto di studi specifici, perciò questo medicamento non deve essere usato in questa fascia di età.
Dosaggio in caso di broncopneumopatia cronica ostruttiva (BPCO):
Adulti a partire da 40 anni di età: 1 dose singola di Relvar Ellipta 92/22 1 volta al giorno.
Non modifichi di propria iniziativa la posologia prescritta e non interrompa da sé il trattamento. Se ritiene che l'azione del medicamento sia troppo debole o troppo forte, ne parli al suo medico o al suo farmacista.
Prima del primo impiego legga attentamente le istruzioni: è molto importante che il medicamento sia usato in modo corretto.
Se involontariamente ha usato una dose di Relvar Ellipta maggiore di quella prescrittale dal suo medico, si rivolga al suo medico o al suo farmacista. Eventualmente noterà un ritmo cardiaco più accelerato del solito, tremori o mal di testa.
Se avesse usato per un periodo prolungato dosi maggiori di quelle prescritte, è molto importante che informi il suo medico o il suo farmacista.
Non utilizzi una quantità doppia per compensare la dimenticanza dell'inalazione precedente. Si limiti ad inalare la dose successiva alla solita ora.
Istruzioni per l'uso
Prima del primo impiego, legga attentamente le Istruzioni per l'uso, perché è molto importante che il medicamento sia usato nel modo corretto.
La confezione dell'inalatore Ellipta contiene le parti seguenti:
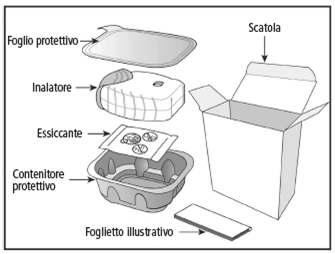
L'inalatore è confezionato in un contenitore protettivo. Lo apra solo quando lei è pronto/a per l'inalazione. Per aprirlo rimuova il foglio protettivo. Nel contenitore è presente anche una bustina di essiccante per ridurre l'umidità. Getti via la bustina di essiccante. La bustina di essiccante non deve essere aperta e il suo contenuto non va deglutito né aspirato.
Quando viene estratto dalla sua confezione, l'inalatore è in posizione chiusa. Apra l'inalatore soltanto per inalare una dose, ma solo quando lei è pronto/a per l'inalazione.
Dopo aver estratto l'inalatore dal contenitore protettivo, annoti sull'etichetta dell'inalatore la data di apertura del contenitore protettivo. Dopo apertura del contenitore protettivo l'inalatore è valido per 6 settimane, dopo le quali non deve essere più usato.
Prima del primo impiego legga attentamente la sezione seguente
Se apre e chiude il cappuccio protettivo senza inalare il medicamento, la dose va persa.
Infatti, la dose rimane nell'inalatore, ma non può più essere inalata.
In questo modo è escluso che inavvertitamente venga inalata in una volta sola una dose troppo elevata o doppia.
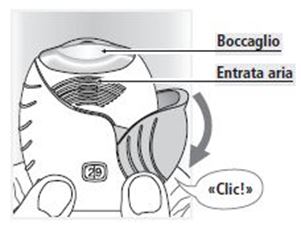
1. Preparazione di una dose
Prima di aprire il cappuccio protettivo aspetti fino a quando è pronto a inalare la dose.
Spinga completamente verso il basso il cappuccio protettivo, fino a sentire un «clic».
A questo punto il medicamento è pronto per l'inalazione.
Il numero indicato dal contadosi diminuisce di 1 unità.
- Eviti assolutamente di agitare l'inalatore.
- Se, malgrado il clic ben udibile, il numero indicato non diminuisce, significa che l'inalatore non eroga il medicamento. In tal caso riporti l'inalatore in farmacia e chieda consiglio al suo farmacista.
2. Inalazione del medicamento
Innanzi tutto espiri il più a fondo possibile, finché lo può fare senza sforzo; durante l'espirazione la bocca deve essere tenuta lontana dall'inalatore.
Non espiri nell'inalatore.
Chiuda bene le labbra intorno al boccaglio.
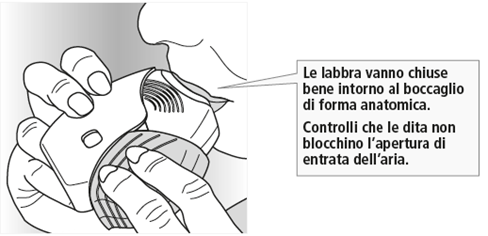
Inspiri profondamente, a lungo e in modo costante. Successivamente, trattenga il respiro il più a lungo possibile (almeno 3 - 4 secondi).
Stacchi l'inalatore dalla bocca.
Espiri lentamente e con calma.
Anche con un uso corretto dell'inalatore può succedere che non senta il gusto né in altro modo noti di avere inalato il medicamento.
3. Chiudere l'inalatore e sciacquare la bocca
Se prima di richiudere il cappuccio dell'inalatore vuole pulire il boccaglio, utilizzi semplicemente un panno asciutto.
Spinga il più possibile verso l'alto il cappuccio dell'inalatore, finché il boccaglio è di nuovo coperto completamente.
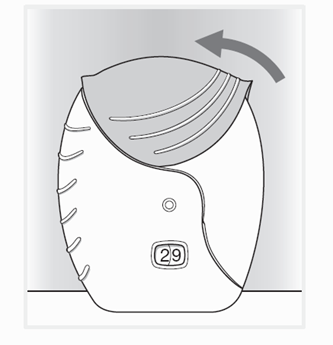
Dopo l'uso dell'inalatore sciacqui bene la bocca con acqua. Questo riduce il rischio di effetti collaterali, come la formazione di ulcere in bocca e in gola.
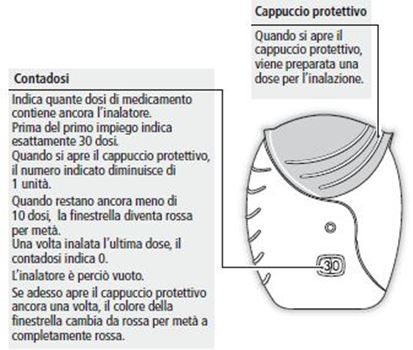
Avvertenze
- Quando la finestrella del contadosi appare rossa per metà, bisogna procurarsi un nuovo inalatore. Quando la finestrella del contadosi è completamente rossa, l'inalatore è vuoto e gliene occorre uno nuovo.
- Non cerchi di manipolare il contadosi né di staccarlo dal corpo dell'inalatore. Il contadosi non può essere riportato indietro né staccato dall'inalatore.
- Se il suo medico le ha dato istruzioni diverse per l'uso del suo inalatore, si attenga scrupolosamente a tali istruzioni. Se avesse problemi con l'uso dell'inalatore, si rivolga al suo medico o al suo farmacista.
Quali effetti collaterali può avere Relvar Ellipta?
Dopo l'uso di Relvar Ellipta è raro il manifestarsi di difficoltà respiratorie improvvise (interessano meno di 1 persona su 1000). Se le difficoltà respiratorie o il respiro sibilante peggiorano immediatamente dopo l'uso di Relvar Ellipta, interrompa subito l'uso del medicamento e consulti al più presto possibile il suo medico.
Con l'uso di Relvar Ellipta possono manifestarsi anche gli effetti collaterali seguenti.
Molto frequentemente (in più di 1 su 10 pazienti) durante il trattamento possono comparire mal di testa o sintomi simili a quelli dell'influenza.
Frequentemente (fino ad 1 su 10 pazienti) si possono sviluppare micosi in bocca e in gola. Può combattere questo effetto collaterale sciacquando immediatamente la bocca con acqua dopo ogni uso di Relvar Ellipta.
Frequentemente sono stati osservati anche infiammazioni delle vie aeree inferiori (bronchite), infezioni e infiammazioni dei seni paranasali o della gola, influenza, dolori e manifestazioni irritative nella regione posteriore della cavità orale e in gola, naso che prude, gocciola o è chiuso, tosse, raucedine e polmonite. La polmonite è più frequente nei pazienti con BPCO che in quelli con asma. Avverta il suo medico se durante il trattamento con Relvar Ellipta nota uno dei seguenti possibili sintomi di polmonite: febbre, brividi, aumento dell'espettorato o cambiamento del suo colore, tosse più frequente o aggravamento delle difficoltà respiratorie.
Frequentemente sono stati riportati anche osteoporosi con rischio di fratture ossee, dolori addominali, dolori alla schiena, dolori articolari, crampi muscolari e febbre.
Occasionalmente (fino ad 1 su 100 pazienti) possono manifestarsi un battito cardiaco irregolare o accelerato come anche delle palpitazioni o aumento della glicemia.
Raramente (fino ad 1 su 1000 pazienti) possono manifestarsi disturbi psichici come iperattività, disturbi del sonno, ansia, depressioni o aggressività.
In casi rari sono state osservate anche reazioni allergiche. Interrompa immediatamente l'uso del medicamento e si rivolga il più presto possibile al suo medico, se dopo l'utilizzo di Relvar osserva uno dei seguenti sintomi di una possibile reazione allergica: eruzione cutanea o arrossamento cutaneo, gonfiori (occasionalmente anche del viso o della bocca, dovuti al cosiddetto angioedema), difficoltà respiratorie improvvise quali affanno respiratorio, respirazione sibilante o tosse, debolezza o stordimento improvviso (che potrebbe portare fino a collasso o perdita di coscienza).
Sono stati osservati anche ansia e tremori.
In caso di sovradosaggio, deve consultare immediatamente il medico.
Se osserva effetti collaterali qui non descritti dovrebbe informare il suo medico o il suo farmacista.
Di che altro occorre tener conto?
Il medicamento non dev'essere utilizzato oltre la data indicata con «EXP» sul contenitore.
Relvar Ellipta va conservato fuori dalla portata dei bambini, a temperatura non superiore a 25 °C e nella confezione originale, per proteggere il contenuto dall'umidità. Il foglio protettivo va tolto solo appena prima del primo impiego dell'inalatore.
Dopo l'apertura del contenitore protettivo il medicamento si conserva per 6 settimane.
Subito dopo aver estratto l'inalatore dal contenitore protettivo, deve annotare sull'etichetta dell'inalatore la data di apertura del contenitore protettivo.
Il medico o il farmacista, che sono in possesso di un'informazione professionale dettagliata, possono darle ulteriori informazioni.
Cosa contiene Relvar Ellipta?
Relvar Ellipta 92/22 contiene, come principi attivi, fluticasone furoato (92 µg per dose erogata) e vilanterolo (22 µg per dose erogata) e, come sostanze ausiliarie, lattosio (con piccole quantità di lattoproteine) e magnesio stearato.
Relvar Ellipta 184/22 contiene, come principi attivi, fluticasone furoato (184 µg per dose erogata) e vilanterolo (22 µg per dose erogata) e, come sostanze ausiliarie, lattosio (con piccole quantità di lattoproteine) e magnesio stearato.
Numero dell’omologazione
62969 (Swissmedic).
Dove è ottenibile Relvar Ellipta? Quali confezioni sono disponibili?
Relvar Ellipta è ottenibile in farmacia, dietro presentazione della prescrizione medica.
Relvar Ellipta 92/22
Confezione con 1 inalatore Ellipta da 30 dosi singole
Confezione con 3 inalatori Ellipta da 30 dosi singole
Relvar Ellipta 184/22
Confezione con 1 inalatore Ellipta da 30 dosi singole
Confezione con 3 inalatori Ellipta da 30 dosi singole
Titolare dell’omologazione
GlaxoSmithKline AG, 3053 Münchenbuchsee.
Questo foglietto illustrativo è stato controllato l'ultima volta nel gennaio 2019 dall'autorità competente in materia di medicamenti (Swissmedic).
Zusammensetzung
Wirkstoffe: Fluticasoni-17 furoas, Vilanterolum (ut Vilanteroli trifenatas).
Hilfsstoffe: Lactosum monohydricum (mit geringen Mengen an Milchprotein), Magnesii stearas, q.s. ad pulverem.
Galenische Form und Wirkstoffmenge pro Einheit
Einzeldosiertes Pulver zur Inhalation
Eine Einzeldosis Relvar Ellipta 92/22 enthält 100 µg Fluticasonfuroat und 25 µg Vilanterol (als Vilanteroltrifenatat). Die abgegebene Dosis (die Dosis, welche vom Mundstück des Ellipta-Inhalators abgegeben wird) beträgt 92 µg Fluticasonfuroat und 22 µg Vilanterol.
Eine Einzeldosis Relvar Ellipta 184/22 enthält 200 µg Fluticasonfuroat und 25 µg Vilanterol (als Vilanteroltrifenatat). Die abgegebene Dosis (die Dosis, welche vom Mundstück des Ellipta-Inhalators abgegeben wird) beträgt 184 µg Fluticasonfuroat und 22 µg Vilanterol.
Ein Ellipta Inhalator enthält 30 Einzeldosen zur Inhalation.
Indikationen/Anwendungsmöglichkeiten
Asthma
Relvar Ellipta wird zur regelmässigen Behandlung von Asthma bronchiale angewendet, wenn ein Kombinationspräparat (ein lang wirksamer Beta-2-Agonist und ein inhalatives Kortikosteroid) angezeigt ist: Bei Erwachsenen und Jugendlichen ab 12 Jahren, die mit inhalativen Kortikosteroiden und bedarfsweise angewendeten, kurzwirksamen inhalativen Beta-2-Agonisten nicht ausreichend eingestellt sind.
COPD
Symptomatische Behandlung von chronisch obstruktiver Lungenkrankheit (COPD) bei Patienten mit einem FEV1 <70% und ≥2 Exazerbationen in den letzten 12 Monaten.
Dosierung/Anwendung
Relvar Ellipta ist ausschliesslich zur (oralen) Inhalation bestimmt.
Relvar Ellipta wird einmal täglich, immer zur gleichen Tageszeit verabreicht.
Die Patienten sollen darauf hingewiesen werden, nach der Inhalation den Mund mit Wasser auszuspülen, ohne das Spülwasser zu schlucken, um Mundsoor und Rachenirritationen zu vermeiden.
Asthma
Erwachsene und Jugendliche ab 12 Jahren:
Eine Inhalation Relvar Ellipta 92/22 1-mal täglich.
oder
Eine Inhalation Relvar Ellipta 184/22 1-mal täglich.
In der Regel ist innerhalb der ersten 15 Minuten nach der Inhalation von Relvar Ellipta eine Verbesserung der Lungenfunktion feststellbar. Allerdings müssen die Patienten darauf hingewiesen werden, dass zur dauerhaften Kontrolle ihrer Asthmasymptome eine regelmässige tägliche Anwendung erforderlich ist und dass die Behandlung auch dann fortgesetzt werden muss, wenn keine Symptome mehr vorhanden sind.
Falls im Zeitraum zwischen zwei Verabreichungen Asthmasymptome auftreten, sollte zur sofortigen Symptomlinderung ein kurzwirkender Beta-2-Agonist eingesetzt werden.
Bei Erwachsenen und bei Jugendlichen ab 12 Jahren, die eine niedrige bis mittlere Dosis eines inhalativen Kortikosteroids in Kombination mit einem langwirkenden Beta-2-Agonisten benötigen, sollte Relvar Ellipta in der Anfangsdosierung von 92/22 µg verordnet werden. Wenn die Patienten unter Relvar Ellipta 92/22 nicht ausreichend kontrolliert sind, kann eine Aufdosierung auf 184/22 µg in Erwägung gezogen werden.
Die Patienten müssen weiterhin regelmässig ärztlich beurteilt werden, damit sie Fluticasonfuroat/Vilanterol stets in der jeweils optimalen Dosisstärke erhalten. Eine Dosisänderung sollte nur auf ärztliche Anweisung erfolgen. Dabei muss immer auf die niedrigste, zur Aufrechterhaltung einer wirksamen Symptomkontrolle ausreichende Dosis eingestellt werden. Der verordnende Arzt sollte dabei berücksichtigen, dass bei Asthma-Patienten Fluticasonfuroat (FF) 92 µg einmal täglich in der Wirkung ungefähr vergleichbar ist mit einer zweimaligen täglichen Verabreichung von Fluticasonpropionat (FP) 250 µg während FF 184 µg einmal täglich vergleichbar ist mit zweimal täglich FP 500 µg.
Kinder unter 12 Jahren:
Die Sicherheit und Wirksamkeit von Relvar Ellipta in Kindern unter 12 Jahren mit Asthma wurde noch nicht genügend untersucht.
COPD
Erwachsene ab 40 Jahren:
Eine Inhalation Relvar Ellipta 92/22 1-mal täglich.
Die Patienten stellen in der Regel innerhalb der ersten 16–17 Minuten nach der Inhalation von Relvar Ellipta eine Verbesserung ihrer Lungenfunktion fest.
Relvar Ellipta 184/22 ist nicht zur Anwendung bei Patienten mit COPD zugelassen. Ein Zusatznutzen von Relvar 184/22 gegenüber Relvar 92/22 konnte für die Anwendung bei COPD nicht gezeigt werden; es besteht ein potentiell erhöhtes Risiko einer Pneumonie und systemischer steroidbedingter unerwünschter Wirkungen (vgl. «Warnhinweise und Vorsichtsmassnahmen»).
Besondere Patientengruppen (Asthma und COPD)
Ältere Patienten (>65 Jahre)
Bei dieser Patientengruppe ist keine Anpassung der Dosierung erforderlich (vgl. «Pharmakokinetik»).
Eingeschränkte Nierenfunktion
Bei dieser Patientengruppe ist keine Anpassung der Dosierung erforderlich (vgl. «Pharmakokinetik»).
Eingeschränkte Leberfunktion
In Studien an Patienten mit leichter, mässiger und hochgradiger Leberfunktionsbeeinträchtigung wurde eine erhöhte systemische Fluticasonfuroat-Exposition (AUC) festgestellt (vgl. «Pharmakokinetik»).
Bei der Verabreichung an Patienten mit eingeschränkter Leberfunktion, bei welchen die Gefahr von kortikosteroidbedingten unerwünschten systemischen Wirkungen unter Umständen höher sein kann, ist Vorsicht geboten. Für Patienten mit mässig oder hochgradig beeinträchtigter Leberfunktion beträgt die zulässige Höchstdosis eine tägliche Inhalation mit Relvar Ellipta 92/22.
Kontraindikationen
Relvar Ellipta ist kontraindiziert bei Patienten mit bekannter Überempfindlichkeit gegenüber einem Inhaltsstoff oder schwerer Milcheiweiss-Allergie (vgl. «Zusammensetzung»).
Warnhinweise und Vorsichtsmassnahmen
Relvar Ellipta darf nicht zur Behandlung akuter Asthmasymptome oder akuter COPD-Exazerbationen angewendet werden, bei denen jeweils ein kurzwirkender Bronchodilatator erforderlich ist. Wenn die Anwendung von kurzwirkenden Bronchodilatatoren zur Symptomlinderung immer häufiger erforderlich wird, ist dies ein Hinweis auf eine Verschlechterung der Symptomkontrolle; in diesem Fall müssen die betreffenden Patienten ärztlich beurteilt werden.
Die Patienten sollten darauf hingewiesen werden, die Behandlung mit Relvar Ellipta nicht ohne ärztliche Überwachung abzubrechen, da die Symptome nach dem Absetzen des Arzneimittels wieder auftreten können.
Unter der Behandlung mit Relvar Ellipta können asthmabedingte unerwünschte Ereignisse und Exazerbationen eintreten. Die Patienten sollten darauf hingewiesen werden, mit der Behandlung fortzufahren, aber ärztliche Hilfe in Anspruch zu nehmen, wenn die Asthmasymptomatik nach Einleitung der Behandlung mit Relvar Ellipta weiterhin unkontrolliert bleibt oder sich verschlechtert.
Paradoxer Bronchospasmus
Wie bei anderen inhalativen Therapien kann es zu einem paradoxen Bronchospasmus mit sofortiger Verstärkung des Giemens nach Verabreichung kommen. Dieser muss umgehend mit einem kurzwirkenden inhalativen Bronchodilatator behandelt werden. In diesem Fall ist Relvar Ellipta unverzüglich abzusetzen; der betreffende Patient muss eingehend untersucht werden und gegebenenfalls eine alternative Behandlung erhalten.
Kardiovaskuläre Effekte
Sympathomimetische Arzneimittel wie Relvar Ellipta können kardiovaskuläre Wirkungen wie Herzrhythmusstörungen, z.B. supraventrikuläre Tachykardie und Extrasystolen, hervorrufen. Ausserdem wurde bei Beta-2-Agonisten wie Relvar Ellipta von elektrokardiographischen Veränderungen berichtet, wie z.B. einem Abflachen der T-Welle, einer Verlängerung des QTc-Intervalls und einer Senkung der ST-Strecke; die klinische Signifikanz dieser Veränderungen ist jedoch unbekannt. Vor der Verschreibung einer Dauerbehandlung mit einem Beta-2-Agonisten, wie Relvar Ellipta, sollten COPD-Patienten hinsichtlich kardiovaskulärer Begleiterkrankungen abgeklärt werden. Dabei empfiehlt sich vorsichtshalber auch die Durchführung einer EKG-Untersuchung mit Frage nach QTc-Verlängerung.
Patienten mit beeinträchtigter Leberfunktion
Bei Patienten mit mässig oder hochgradig beeinträchtigter Leberfunktion sollte Relvar Ellipta 92/22 angewendet werden und diese Patienten sollten hinsichtlich des Auftretens von kortikosteroidbedingten unerwünschten systemischen Wirkungen überwacht werden (vgl. «Dosierung/Anwendung»).
Systemische Kortikosteroideffekte
Die gleichzeitige Gabe von Relvar mit starken Inhibitoren von CYP3A4 sollte vermieden werden.
Systemische Wirkungen können unter allen inhalativen Kortikosteroiden auftreten, insbesondere, wenn sie langfristig in hohen Dosen verschrieben werden. Die Wahrscheinlichkeit derartiger Wirkungen ist jedoch geringer als bei oralen Kortikosteroiden. Zu den möglichen systemischen Wirkungen gehören eine Suppression der Hypothalamus-Hypophysen-Nebennieren-(HPA-)Achse, eine Abnahme der Knochendichte sowie Katarakt und Glaukom, oder seltene Erkrankungen, wie zentrale seröse Chorioretinopathie (CSCR). Selten können inhalative Kortikosteroide mit psychischen Störungen einhergehen, einschliesslich psychomotorischer Hyperaktivität, Schlafstörungen, Angst, Depression oder Aggression (insbesondere bei Kindern).
Sehstörungen:
Bei der systemischen und topischen Anwendung von Kortikosteroiden können Sehstörungen auftreten. Wenn ein Patient mit Symptomen wie verschwommenem Sehen oder anderen Sehstörungen vorstellig wird, sollte eine Überweisung des Patienten an einen Augenarzt zur Bewertung möglicher Ursachen in Erwägung gezogen werden; diese umfassen unter anderem Katarakt, Glaukom oder seltene Erkrankungen, wie z.B. CSCR, die nach der Anwendung systemischer oder topischer Kortikosteroide gemeldet wurden.
Wie alle kortikoidhaltigen Arzneimittel darf Relvar Ellipta an Patienten mit Lungentuberkulose oder mit chronischen bzw. unbehandelten Infektionen nur bei Einhaltung entsprechender Vorsichtsmassnahmen verabreicht werden.
Hyperglykämie
Unter Relvar Ellipta wurde über erhöhte Blutzuckerspiegel bei Diabetespatienten berichtet. Bei einer Verschreibung von Relvar Ellipta an Patienten mit Diabetes sollte dies berücksichtigt werden.
Pneumonien
Bei COPD-Patienten unter Relvar Ellipta traten gehäuft Fälle von Pneumonie auf. Auch die Inzidenz hospitalisierungsbedürftiger Pneumonien war erhöht. In manchen Fällen verliefen solche Pneumonien auch tödlich (vgl. «Unerwünschte Wirkungen»). Von ärztlicher Seite muss bei COPD-Patienten auf die mögliche Entwicklung einer Pneumonie geachtet werden, da die klinischen Merkmale solcher Infektionen sich mit den Symptomen einer COPD-Exazerbation teilweise decken. Risikofaktoren für eine Pneumonie bei COPD-Patienten unter Relvar Ellipta sind Rauchen, anamnestisch bekannte Pneumonie, Body-Mass-Index <25 kg/m2 und ein forciertes Exspirationsvolumen in der ersten Sekunde (FEV1) von <50% des Sollwerts. Diese Faktoren sind bei der Verschreibung von Relvar Ellipta zu berücksichtigen; beim Auftreten einer Pneumonie muss eine Neubeurteilung der Behandlung erfolgen.
Relvar Ellipta 184/22 ist nicht zur Anwendung bei Patienten mit COPD zugelassen. Ein Zusatznutzen von Relvar Ellipta 184/22 gegenüber Relvar Ellipta 92/22 konnte für die Anwendung bei COPD nicht gezeigt werden; es besteht ein potentiell erhöhtes Risiko einer Pneumonie und systemischer steroidbedingter unerwünschter Wirkungen (vgl. «Unerwünschte Wirkungen»).
Das Auftreten von Pneumonien in Asthmapatienten war unter Relvar 184/22 häufig. Unter Relvar 184/22 traten numerisch häufiger Pneumonien auf als unter Relvar 92/22 oder Plazebo (vgl. «Unerwünschte Wirkungen»). Es konnten keine Risikofaktoren identifiziert werden.
Hilfsstoffe
Relvar Ellipta enthält Laktose. Patienten mit einer seltenen erblichen Form von Galaktose-Intoleranz, Laktase-Mangel oder Glukose-Galaktose Malabsorption sollten Relvar Ellipta nicht anwenden.
Interaktionen
Interaktionssstudien wurden nur an Erwachsenen durchgeführt.
Pharmakokinetische Interaktionen:
Beta-Blocker
Beta-adrenerge Blocker können die Wirkung von Beta-2-adrenergen Agonisten abschwächen oder antagonisieren. Die gleichzeitige Anwendung nichtselektiver bzw. selektiver Beta-adrenerger Blocker muss vermieden werden, sofern ihr Einsatz nicht zwingend indiziert ist.
CYP3A4-Inhibitoren
Sowohl Fluticasonfuroat als auch Vilanterol werden im Rahmen eines umfassenden, durch das Leberenzym CYP3A4 vermittelten First-Pass-Metabolismus rasch abgebaut.
Die gleichzeitige Verabreichung mit starken CYP3A4 Inhibitoren (z.B. Ketoconazol, Itraconazol, Clarithromycin, Ritonavir oder Produkte, welche Cobicistat enthalten) kann zu erhöhter Exposition gegenüber Kortikosteroiden als auch gegenüber Vilanterol führen und somit zu einem erhöhten Risiko von Nebenwirkungen, insbesondere systemischer Kortikosteroide. Die gleichzeitige Verabreichung sollte vermieden werden, es sei denn, der Nutzen überwiegt das erhöhte Risiko systemischer Kortikosteroid-Nebenwirkungen; in diesem Fall sollten die Patienten im Hinblick auf systemische Kortikosteroid-Nebenwirkungen überwacht werden (vgl. «Pharmakokinetik»).
P-Glycoprotein-Inhibitoren
Sowohl Fluticasonfuroat als auch Vilanterol sind Substrate des P-Glycoproteins (P-gp). Eine Studie zur klinischen Pharmakologie, bei der Vilanterol zusammen mit Verapamil, einem starken P-gp- und moderaten CYP3A4-Inhibitor, an gesunde Personen verabreicht wurde, ergab keinen relevanten Einfluss auf die Pharmakokinetik von Vilanterol. Studien zur klinischen Pharmakologie mit einem spezifischen P-gp-Hemmer und Fluticasonfuroat wurden nicht durchgeführt.
Pharmakodynamische Interaktionen:
Arzneimittel, die bekanntermassen eine Verlängerung des QTc-Intervalls bewirken
Wie bei anderen Beta-2-Agonisten besteht theoretisch das Risiko, dass Arzneimittel, die bekanntermassen das QTC-Intervall verlängern können, zu einer pharmakodynamischen Interaktion mit dem in Relvar Ellipta enthaltenen Vilanterol führen und das mögliche Risiko von ventrikulären Arrhythmien erhöhen können. Beispiele solcher Arzneimittel sind bestimmte Antihistaminika (z.B. Terfenadin, Mizolastin), gewisse Antiarrhythmika (z.B. Chinidin), Phenothiazine, Erythromycin, Ritonavir und trizyklische Antidepressiva. Die zusätzliche Verabreichung sympathikomimetischer Substanzen kann unerwünschte kardiovaskuläre Wirkungen verstärken. Wird Relvar Ellipta an Patienten verabreicht, die unter Behandlung mit MAO-Hemmern oder trizyklischen Antidepressiva stehen, dann sollte dies mit Vorsicht geschehen, da die Wirkung von Beta-2-Stimulatoren auf das kardiovaskuläre System verstärkt werden kann.
Die gleichzeitige Verabreichung von L-Dopa, L-Thyroxin und Oxytocin kann die kardiale Toleranz gegenüber Beta-2-Sympathomimetika negativ beeinflussen.
Sympathomimetika
Die gleichzeitige Anwendung anderer Sympathomimetika (alleine oder als Teil einer Kombinationstherapie) könnte die unerwünschten Wirkungen von Vilanterol verstärken.
Hypokaliämie
Bei gleichzeitiger Behandlung mit Methylxanthin-Derivaten, Steroiden oder nicht kaliumsparenden Diuretika könnte sich eine durch beta2-adrenerge Agonisten möglicherweise hervorgerufene Hypokaliämie verstärken.
Schwangerschaft/Stillzeit
Schwangerschaft
Zur Exposition in der Schwangerschaft liegt beim Menschen nur eingeschränktes Datenmaterial vor.
Aus tierexperimentellen Studien ist eine Reproduktionstoxizität nach Verabreichung von Beta-2-Agonisten und Kortikosteroiden bekannt (vgl. «Präklinische Daten»).
Relvar Ellipta darf während der Schwangerschaft nicht angewendet werden, es sei denn es ist klar notwendig.
Stillzeit
Zur Ausscheidung von Fluticasonfuroat oder Vilanterol bzw. ihren Metaboliten in die Muttermilch liegt nur eingeschränktes Datenmaterial vor. Andere Kortikosteroide und Beta-2-Agonisten sind jedoch in der Muttermilch nachweisbar.
Ein Risiko für gestillte Neugeborene/Säuglinge kann nicht ausgeschlossen werden.
Unter Abwägung der Vorteile des Stillens für das Kind und des Nutzens der Behandlung für die Mutter muss entweder abgestillt oder die Behandlung mit Relvar Ellipta eingestellt werden.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von Maschinen
Es wurden keine spezifischen Studien zum Einfluss von Relvar Ellipta auf die Fahrtüchtigkeit und die Fähigkeit zum Bedienen von Maschinen durchgeführt.
Unerwünschte Wirkungen
Die Häufigkeit unerwünschter Wirkungen im Zusammenhang mit Relvar Ellipta wurde anhand von Daten aus grossangelegten klinischen Studien zu Asthma und COPD bestimmt. Im klinischen Entwicklungsprogramm für Asthma gingen die Daten von insgesamt 7'034 Patienten in eine integrierte Auswertung der unerwünschten Wirkungen ein. Im klinischen Entwicklungsprogramm für COPD gingen die Daten von insgesamt 6'237 Patienten in eine integrierte Auswertung der unerwünschten Reaktionen ein.
Mit Ausnahme von Pneumonie und Frakturen fiel das Sicherheitsprofil bei Patienten mit Asthma und COPD ähnlich aus. In den klinischen Studien wurden Pneumonie und Frakturen bei Patienten mit COPD häufiger beobachtet.
Die unerwünschten Wirkungen sind nach Systemorganklasse und Häufigkeit aufgelistet. Die Häufigkeiten sind gemäss der folgenden Konvention absteigend geordnet: sehr häufig (>1/10), häufig (≥1/100 bis <1/10), gelegentlich (≥1/1'000 bis <1/100), selten (≥1/10'000 bis <1/1'000), sehr selten (<1/10'000).
Innerhalb jeder Häufigkeitskategorie sind die unerwünschten Reaktionen absteigend nach ihrem Schweregrad aufgeführt.
Infektionen und Infestationen
Häufig: Pneumonie*, Infektionen der oberen Atemwege, Bronchitis, Grippesymptome, Candidose von Mund und Rachen.
Nervensystem
Sehr häufig: Kopfschmerzen (12–17%).
Herz
Gelegentlich: Extrasystolen.
Atmungsorgane
Sehr häufig: Nasopharyngitis (10–14%).
Häufig: Schmerzen im Mund- und Rachenraum, Sinusitis, Pharyngitis, Rhinitis, Husten, Heiserkeit.
Gastrointestinale Störungen
Häufig: Bauchschmerzen.
Muskelskelettsystem
Häufig: Gelenkschmerzen, Rückenschmerzen, Frakturen**.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Pyrexie.
Post-Marketing Daten:
Erkrankungen des Immunsystems
Selten: Hypersensitivitätsreaktionen einschliesslich Anaphylaxie, Angioödem, Rash und Urtikaria.
Stoffwechsel- und Ernährungsstörungen
Gelegentlich: Hyperglykämie.
Psychiatrische Erkrankungen
Unbekannt: Angst.
Nervensystem
Unbekannt: Tremor.
Herz
Gelegentlich: Palpitationen, Tachykardie.
Muskelskelettsystem
Häufig: Muskelkrämpfe.
Atmungsorgane
Selten: paradoxer Bronchospasmus.
Beschreibung ausgewählter unerwünschter Ereignisse
* Pneumonie (s. «Warnhinweise und Vorsichtsmassnahmen»)
In zwei 12-monatigen Studien an insgesamt 3‘255 COPD-Patienten (mittlere post-bronchodilatorische FEV1 45% des vorhergesehenen Wertes, Standardabweichung 13%), die im vorangegangenen Jahr eine COPD-Exazerbation durchgemacht hatten, war die Pneumonie-Inzidenz bei Patienten unter der Kombination Fluticasonfuroat (FF, in den Dosisstärken 46, 92 und 184 µg)/Vilanterol (VI) 22 µg höher (6%–7%) als bei Patienten, die ausschliesslich VI 22 µg erhielten (3%). Eine hospitalisierungsbedürftige Pneumonie trat bei 3% der Patienten unter FF/VI (alle Dosisstärken) und bei <1% der Patienten unter VI auf. Aus diesen Studien wurden neun Pneumoniefälle mit tödlichem Verlauf gemeldet. Davon traten sieben unter der Behandlung mit FF/VI 184/22 auf, einer unter der Behandlung mit FF/VI 92/22 und einer nach Abschluss der VI-Monotherapie.
In SUMMIT, einer randomisierten Multizenterstudie (HZC113782) zur Untersuchung der Gesamt-Mortalität wurden 16'568 Teilnehmer nebst Bronchodilatatoren nach Bedarf zusätzlich mit FF/VI 92/22, FF 92, VI 22 oder Placebo über durchschnittlich 1,7 Jahre behandelt. Die Teilnehmer litten an moderater COPD (mittlere post-bronchodilatorische FEV1 60% des vorhergesehenen Wertes, SD 6%). Die meisten Patienten hatten <2 Exazerbationen in den letzten 12 Monaten (GOLD-Risiko-Gruppe B), weshalb eine Übertragung von Resultaten betreffend Überleben und Sicherheit auf die in der Schweiz zugelassene Zielpopulation (GOLD-Risiko-Gruppe D) nur beschränkt möglich ist.
Die unerwünschten Ereignisse Pneumonie sind in nachfolgender Tabelle dargestellt.
| Ereignisse unter Behandlung | Anzahl (%) Patienten [Ereignisrate pro 1000 Behandlungsjahre] | |||
| FF/VI 92/22 N=4140 | FF 92 N=4157 | VI 22 N=4140 | Placebo N=4131 | |
| Pneumonie | 237 (6) [39.5] | 228 (5) [42.4] | 163 (4) [27.7] | 214 (5) [38.4] |
| Schwere Pneumonie | 140 (3) [22.4] | 146 (4) [25.1] | 104 (3) [16.4] | 127 (3) [22.2] |
| Todesfälle infolge Pneumonie | 13 (<1) [1.8] | 10 (<1) [1.5] | 6 (<1) [0.9] | 9 (<1) [1.4] |
In einer integrierten Analyse der Daten aus 11 Studien zu Asthma (7‘034 Patienten) wurde unter FF/VI 92/22 eine placeboähnliche Pneumonie-Inzidenz (expositionskorrigiert aufgrund der geringen Fallzahlen und der eingeschränkten Zahl an Placebo-Patienten) beobachtet (9,6/1‘000 Patientenjahre unter FF/VI, 8,0/1‘000 Patientenjahre unter Placebo). Die Pneumonie-Inzidenz war unter FF/VI 184/22 höher (18,4/1‘000 Patientenjahre) als bei der 92/22-µg-Dosisstärke. Unter beiden Dosisstärken war nur bei wenigen Pneumonie-Ereignissen eine Hospitalisierung erforderlich; Unterschiede bezüglich der Inzidenz schwerwiegender Ereignisse wurden zwischen den beiden Dosisstärken nicht beobachtet.
** Frakturen
In zwei 12-monatigen Wiederholungsstudien an insgesamt 3‘255 COPD-Patienten war die Gesamtinzidenz von Knochenfrakturen in allen Behandlungsgruppen niedrig. Die Inzidenz fiel dabei in den FF/VI-Gruppen höher aus (2%) als in der Gruppe mit VI 22 µg (<1%). Obwohl in den Gruppen mit FF/VI im Vergleich zur Gruppe mit VI 22 µg mehr Frakturen auftraten, wurden die unter Kortikoidanwendung typischen Frakturen (z.B. Rückenmarkskompression/thorakolumbale Wirbelfrakturen, Hüft- und Acetabulumfrakturen) in den Behandlungsarmen mit FF/VI und VI bei <1% der Patienten festgestellt.
Frakturen der SUMMIT Studie (Beschreibung s. oben) sind in der Tabelle aufgelistet:
| Ereignisse unter Behandlung | Anzahl (%) Patienten [Ereignisrate pro 1000 Behandlungsjahre] | |||
| FF/VI 92/22 N=4140 | FF 92 N=4157 | VI 22 N=4140 | Placebo N=4131 | |
| Alle Frakturen | 82 (2) [13.6] | 66 (2) [12.8] | 74 (2) [13.2] | 69 (2) [11.5] |
| Frakturen häufig in Verbindung mit ICS Gebrauch | 23 (<1) [3.4] | 24 (<1) [3.9] | 17 (<1) [2.4] | 13 (<1) [2.1] |
In einer integrierten Analyse der Daten aus 11 Studien zu Asthma (7‘034 Patienten) traten Frakturen mit einer Inzidenz von <1% auf und waren in der Regel mit einem Trauma assoziiert.
Überdosierung
Anzeichen und Symptome
Es liegen keine Daten aus klinischen Studien hinsichtlich einer Überdosierung von Relvar Ellipta vor.
Im Falle einer Überdosierung von Relvar Ellipta kann es zu Anzeichen und Symptomen kommen, die den Wirkungen der einzelnen Komponenten entsprechen, einschliesslich der Wirkungen, die bei einer Überdosierung anderer Beta-2-Agonisten beobachtet werden, sowie der bekannten Klasseneffekte der inhalativen Kortikosteroide (vgl. «Warnhinweise und Vorsichtsmassnahmen»).
Behandlung
Für die Überdosierung von Relvar Ellipta ist keine spezifische Behandlung verfügbar. Im Falle einer Überdosierung muss der Patient die jeweils angemessene unterstützende Behandlung erhalten und entsprechend überwacht werden.
Die kardioselektive β-Blockade kommt nur bei einer schweren Vilanterol-Überdosierung mit ernsten klinischen Symptomen in Betracht, die nicht auf unterstützende Massnahmen anspricht. Bei Patienten mit anamnestisch bekannten Bronchospasmen ist beim Einsatz kardioselektiver β-Blocker Vorsicht geboten.
Das weitere Vorgehen richtet sich nach den klinischen Erfordernissen bzw., sofern zutreffend, nach den Empfehlungen des jeweiligen toxikologischen Informationszentrums.
Eigenschaften/Wirkungen
ATC-Code: R03AK10
Wirkmechanismus
Fluticasonfuroat und Vilanterol sind Vertreter zweier unterschiedlicher Arzneimittelklassen (synthetisches Kortikosteroid bzw. selektiver, langwirkender Beta-2-Rezeptoragonist).
Pharmakodynamische Wirkungen
Fluticasonfuroat (FF)
FF ist ein synthetisches, trifluoriniertes Kortikosteroid mit hochpotenter antiinflammatorischer Wirksamkeit. Der genaue Mechanismus, über den FF die Asthma- und COPD-Symptomatik beeinflusst, ist nicht bekannt. Kortikosteroide haben nachweislich ein breites Wirkungsspektrum auf unterschiedliche Zellarten (z.B. Eosinophile, Makrophagen, Lymphozyten) und Mediatoren (z.B. die am Entzündungsgeschehen beteiligten Zytokine und Chemokine).
Vilanterol Trifenatat (VI)
VI ist ein selektiver, langwirkender Beta-2-adrenerger Agonist (LABA).
Die pharmakologischen Wirkungen von Beta-2-Adrenorezeptoragonisten wie VI beruhen zumindest teilweise auf der Stimulation der intrazellulären Adenylatzyklase, dem Enzym, das die Umwandlung von Adenosintriphosphat (ATP) zu zyklischem Adenosin-3′,5′-monophosphat (cAMP) katalysiert. Erhöhte cAMP-Konzentrationen bewirken eine Entspannung der glatten Bronchialmuskulatur und hemmen die Freisetzung von Mediatoren der Überempfindlichkeitsreaktionen vom Soforttyp, insbesondere aus Mastzellen.
Klinische Wirksamkeit
Asthma
Drei randomisierte, doppelblinde Phase-III-Studien (HZA106827, HZA106829 und HZA106837) unterschiedlicher Dauer untersuchten die Sicherheit und Wirksamkeit von Relvar Ellipta (Fluticasonfuroat/Vilanterol, FF/VI) bei erwachsenen und jugendlichen Patienten mit persistierendem Asthma. Alle Teilnehmer hatten vor Visite 1 mindestens 12 Wochen lang ein ICS (inhalatives Kortikosteroid) mit oder ohne LABA verwendet. Alle Teilnehmer der Studie HZA106837 hatten im Jahr vor Visite 1 mindestens eine Exazerbation durchgemacht, die eine Behandlung mit oralen Kortikosteroiden erforderte. HZA106827 war eine 12-wöchige Studie zur Beurteilung der Wirksamkeit von FF/VI 92/22 µg (n= 201) und FF 92 (n= 205) gegenüber Placebo (n= 203), bei jeweils 1-mal täglicher Verabreichung. HZA106829 war eine 24-wöchige Studie zur Beurteilung der Wirksamkeit von FF/VI 184/22 µg (n= 197) und FF 184 (n= 194), jeweils einmal täglich, gegenüber Fluticasonpropionat 500 µg (FP 500) zweimal täglich (n= 195).
In HZA106827/HZA106829 wurden als ko-primäre Endpunkte die Änderung des Talspiegel-FEV1 (vor Anwendung des Bronchodilatators bzw. der Studienmedikation) bei den Klinikvisiten von Studienbeginn bis zum Ende der Behandlungsphase bei allen Teilnehmern sowie der gewichtete Mittelwert der FEV1-Messreihe 0–24 Stunden nach der Verabreichung bei einer Untergruppe der Teilnehmer am Ende der Behandlungsphase herangezogen. Die Änderung der prozentualen bedarfsmedikationsfreien 24-Stunden-Zeiträume während der Behandlungsphase gegenüber Studienbeginn war ein sekundärer Endpunkt, für den eine ausreichende statistische Trennschärfe gegeben war. Die Ergebnisse für die primären sowie die wichtigsten sekundären Endpunkte dieser Studien sind in Tabelle 1 zusammengefasst.
Tabelle 1: Ergebnisse für die primären und die wichtigsten sekundären Endpunkte der Studien HZA106827 und HZA106829
| Studien-Nr. | HZA106829 | HZA106827 | |||
|---|---|---|---|---|---|
| FF/VI*-Behandlungsdosis (µg) | FF/VI 184/22 einmal täglich vs. FF 184 einmal täglich | FF/VI 184/22 einmal täglich vs. FP 500 zweimal täglich | FF/VI 92/22 einmal täglich vs. FF 92 einmal täglich | FF/VI 92/22 einmal täglich vs. Placebo einmal täglich | |
Änderung des FEV1 bei Talspiegel gegenüber Studienbeginn, LOCF-Ansatz (Übertragung des jeweils letzten Werts) | |||||
| Behandlungsunterschied | 193 ml | 210 ml | 36 ml | 172 ml | |
| p-Wert | p <0,001 | p <0,001 | p= 0,405 | p <0,001 | |
| (95%-KI) | (108; 277) | (127; 294) | (-48; 120) | (87; 258) | |
Gewichteter Mittelwert der FEV1-Messreihe 0–24 h nach Verabreichung | |||||
| Behandlungsunterschied | 136 ml | 206 ml | 116 ml | 302 ml | |
| p-Wert | p= 0,048 | p= 0,003 | p= 0,06 | p <0,001 | |
| (95%-KI) | (1; 270) | (73; 339) | (-5; 236) | (178; 426) | |
Änderung der prozentualen bedarfsmedikationsfreien 24-Stunden-Zeiträume gegenüber Studienbeginn | |||||
| Behandlungsunterschied | 11,7% | 6,3% | 10,6% | 19,3% | |
| p-Wert | p <0,001 | p= 0,067 | p <0,001 | p <0,001 | |
| (95%-KI) | (4,9; 18,4) | (-0,4; 13,1) | (4,3; 16,8) | (13,0; 25,6) | |
Änderung der prozentualen symptomfreien 24-Stunden-Zeiträume gegenüber Studienbeginn | |||||
| Behandlungsunterschied | 8,4% | 4,9% | 12,1% | 18,0% | |
| p-Wert | p= 0,010 | p= 0,137 | p <0,001 | p <0,001 | |
| (95%-KI) | (2,0; 14,8) | (-1,6; 11,3) | (6,2; 18,1) | (12,0; 23,9) | |
Änderung des morgendlichen exspiratorischen Spitzenflusses gegenüber Studienbeginn | |||||
| Behandlungsunterschied | 33,5 l/min | 32,9 l/min | 14,6 l/min | 33,3 l/min | |
| p-Wert | p <0,001 | p <0,001 | p <0,001 | p <0,001 | |
| (95%-KI) | (25,3; 41,7) | (24,8; 41,1) | (7,9; 21,3) | (26,5; 40,0) | |
Änderung des abendlichen exspiratorischen Spitzenflusses gegenüber Studienbeginn | |||||
| Behandlungsunterschied | 30,7 l/min | 26,2 l/min | 12,3 l/min | 28,2 l/min | |
| p-Wert | p <0,001 | p <0,001 | p <0,001 | p <0,001 | |
| (95%-KI) | (22,5; 38,9) | (18,0; 34,3) | (5,8; 18,8) | (21,7; 34,8) | |
* FF/VI = Fluticasonfuroat/Vilanterol = Relvar Ellipta
In HZA106837 war die Behandlungsdauer variabel (mindestens 24 Wochen bis maximal 76 Wochen; die Mehrzahl der Patienten erhielt eine mindestens 52-wöchige Behandlung). In HZA106837 erhielten die Patienten per Randomisierung einmal täglich entweder FF/VI 92/22 (n= 1'009) oder FF 92 (n= 1'010). Primärer Endpunkt war in der Studie HZA106837 die Zeit bis zur ersten schweren Asthma-Exazerbation. Eine schwere Asthma-Exazerbation war definiert als Verschlechterung der Asthma-Symptomatik, bei welcher ein mindestens dreitägiger Einsatz systemischer Kortikosteroide oder eine stationäre Aufnahme bzw. ein Besuch in der Krankenhaus-Notaufnahme zur systemischen Asthma-Behandlung erforderlich war. Die korrigierte mittlere Änderung des Talspiegel-FEV1 gegenüber Studienbeginn wurde als sekundärer Endpunkt ebenfalls ausgewertet.
In HZA106837 war das Risiko einer schweren Asthma-Exazerbation bei Patienten unter FF/VI 92/22 gegenüber Patienten unter FF 92 alleine um 20% reduziert (Hazard-Ratio 0,795; p= 0,036; 95%-KI: 0,642; 0,985). Die Häufigkeit schwerer Asthma-Exazerbationen pro Patient und Jahr betrug in der FF 92-Gruppe 0,19 (ca. 1 alle 5 Jahre) und in der Gruppe mit FF/VI 92/22 0,14 (ca. 1 alle 7 Jahre). Das Verhältnis zwischen den Exazerbationsraten für FF/VI 92/22 und FF 92 betrug 0,755 (95%-KI: 0,603; 0,945). Die Häufigkeit schwerer Asthma-Exazerbationen war damit bei Teilnehmern unter FF/VI 92/22 gegenüber Teilnehmern unter FF 92 um 25% reduziert (p= 0,014). Die 24 Stunden anhaltende bronchodilatatorische Wirkung von Relvar Ellipta blieb bis zum Ende einer einjährigen Behandlungsphase ohne Anzeichen eines Wirksamkeitsverlusts (Tachyphylaxie) bestehen. Unter FF/VI 92/22 wurde im Vergleich zu FF 92 in den Wochen 12, 36 und 52 sowie beim Endpunkt eine einheitliche Verbesserung des Talspiegel-FEV1 um 83 ml bis 95 ml (p <0,001; 95%-KI: 52; 126 ml beim Endpunkt) festgestellt. In der Gruppe mit FF/VI 92/22 wiesen bei Behandlungsende 40% der Patienten eine gute Symptomkontrolle auf (ACQ7 ≤0,75), gegenüber 36% in der Gruppe mit FF 92 (p <0,001; 95%-KI: 1,23; 1,82).
Vergleichsstudien mit Fluticasonpropionat/Salmeterol(FP/Sal)
In einer 24-wöchigen Studie (HZA113091) an erwachsenen und jugendlichen Patienten mit persistierendem Asthma führten sowohl FF/VI 92/22 einmal täglich abends als auch FP/Sal 250/50 µg zweimal täglich zu einer Verbesserung der Lungenfunktion gegenüber Studienbeginn. Die korrigierten mittleren Anstiege des gewichteten Mittelwerts für das 0–24-Stunden-FEV1 gegenüber Studienbeginn von 341 ml (FF/VI 92/22) bzw. 377 ml (FP/Sal 250/50) zeigten eine allgemeine Verbesserung der Lungenfunktion über 24 Stunden unter beiden Behandlungen. Der korrigierte mittlere Behandlungsunterschied von 37 ml zwischen den Gruppen war statistisch nicht signifikant (p= 0,162). Beim FEV1-Talwert erreichten die Patienten in der FF/VI-Gruppe eine mittlere Änderung gegenüber dem Ausgangswert von 281 ml und die Patienten in der FP/Sal-Gruppe eine Änderung von 300 ml (der Unterschied des adjustierten Mittelwerts von 19 ml (95%‑KI: -0,073; 0,034) war statistisch nicht signifikant (p= 0,485)).
Es wurden keine geeigneten Vergleichsstudien versus FP/Sal oder versus andere ICS/LABA Kombinationen durchgeführt, um die Wirkung auf Asthma-Exazerbationen zu vergleichen.
Fluticasonfuroat-Monotherapie
Eine 24-wöchige randomisierte, doppelblinde, placebokontrollierte Studie (FFA112059) untersuchte die Sicherheit und Wirksamkeit von FF 92 einmal täglich (n= 114) sowie von FP 250 zweimal täglich (n= 114) gegenüber Placebo (n= 115) bei erwachsenen und jugendlichen Patienten mit persistierendem Asthma. Alle Teilnehmer mussten bei Visite 1 (Voruntersuchung) seit mindestens 4 Wochen unter Behandlung mit einem ICS in fester Dosierung sein; die Anwendung von LABAs war in den 4 Wochen vor Visite 1 nicht zulässig. Primärer Wirksamkeitsendpunkt war die Änderung des im Rahmen der Klinikvisiten gemessenen FEV1 bei Talspiegel (vor Bronchodilatator und vor Dosisverabreichung) am Ende der Behandlungsphase gegenüber Studienbeginn. Die Änderung der prozentualen bedarfsmedikationsfreien 24-Stunden-Zeiträume während der 24-wöchigen Behandlungsphase gegenüber Studienbeginn war ein sekundärer Endpunkt, für den eine ausreichende statistische Trennschärfe gegeben war. Zum Erhebungszeitpunkt nach 24 Wochen war unter FF und FP ein Anstieg des Talspiegel-FEV1 um 146 ml (95%-KI: 36; 257 ml; p= 0,009) bzw. 145 ml (95%-KI: 33; 257 ml; P= 0,011) gegenüber Placebo feststellbar. Sowohl FF als auch FP führten zu einer Erhöhung der prozentualen bedarfsmedikationsfreien 24-Stunden-Zeiträume um 14,8% (95%-KI: 6,9; 22,7; p <0,001) bzw. 17,9% (95%-KI: 10,0; 25,7; p <0,001) gegenüber Placebo.
Allergen-Provokationsstudie
Die bronchoprotektive Wirkung von FF/VI 92/22 im Hinblick auf asthmatische Reaktionen vom Sofort- und Spättyp auf ein Inhalationsallergen wurde in einer placebokontrollierten Crossover-Studie mit wiederholter Gabe und 4 Behandlungsvarianten (HZA113126) an Patienten mit leichtem Asthma untersucht. Die Patienten erhielten per Randomisierung 21 Tage lang einmal täglich entweder FF/VI 92/22, FF 92, VI 22 oder Placebo. Eine Stunde nach Verabreichung der letzten Dosis wurde dann ein Allergen-Provokationstest durchgeführt. Als Allergen wurden – in Abhängigkeit vom individuellen Testergebnis – Hausstaubmilben, Katzen-Schuppen oder Birkenpollen verwendet. Die Werte von FEV1-Messreihen wurden mit vor der Allergen-Provokation nach einer Inhalation von Kochsalzlösung gemessenen Werten verglichen. Im Vergleich mit FF 92 oder VI 22 alleine wurden die insgesamt grössten Wirkungen auf die asthmatische Reaktion vom Soforttyp unter FF/VI 92/22 beobachtet. Sowohl FF/VI 92/22 als auch FF 92 verhinderten praktisch die asthmatische Reaktion vom Spättyp im Vergleich zur Vilanterol-Monotherapie. Im Metacholin-Provokationstest an Tag 22 bot FF/VI 92/22 einen signifikant stärkeren Schutz vor allergeninduzierter bronchialer Hyperreaktivität als die Monotherapie mit FF bzw. VI.
COPD
Das klinische Entwicklungsprogramm für COPD umfasste eine 12-wöchige (HZC113107), zwei 6-monatige (HZC112206, HZC112207) und zwei einjährige randomisierte, kontrollierte Studien (HZC102970, HZC102871) an Patienten mit klinisch diagnostizierter COPD. In den Studien wurden unter anderem Lungenfunktion, Dyspnoe und die Rate von mittelschweren bis schweren Exazerbationen untersucht.
Sechsmonatige Studien
HZC112206 und HZC112207 waren 24-wöchige, randomisierte, doppelblinde, placebokontrollierte Parallelgruppenstudien, in denen die Wirkung des Kombinationspräparats mit VI bzw. FF alleine und Placebo verglichen wurde. In HZC112206 wurde die Wirksamkeit von FF/VI 46/22 (n= 206) und FF/VI 92/22 (n= 206) mit FF 92 (n= 206), VI 22 (n= 205) und Placebo (n= 207), bei jeweils einmaliger täglicher Verabreichung, verglichen. In HZC112207 wurde die Wirksamkeit von FF/VI 92/22 (n= 204) und FF/VI 184/22 (n= 205) mit FF 92 (n= 204), FF 184 (n= 203) und VI 22 (n= 203) sowie Placebo (n= 205), bei jeweils einmaliger täglicher Verabreichung, verglichen.
Alle Patienten mussten bei der Voruntersuchung eine Raucheranamnese von mindestens zehn Packungsjahren aufweisen, ein FEV1/FVC-Verhältnis nach Salbutamol-Gabe von höchstens 0,70, ein FEV1 nach Salbutamol von höchstens 70% des Sollwerts sowie einen Dyspnoe-Score von ≥2 auf der modifizierten MRC-Skala (Modified Medical Research Council, mMRC; Skala von 0 bis 4). In der Voruntersuchung für die Studien HZC112206 und HZC112207 betrug das durchschnittliche FEV1 vor Bronchodilatator 42,6% bzw. 43,6% des Sollwerts und die mittlere Reversibilität 15,9% bzw. 12,0%. Als ko-primäre Endpunkte wurden in beiden Studien der gewichtete FEV1-Mittelwert von 0 bis 4 Stunden nach der Verabreichung an Tag 168 und die Änderung des Talspiegel-FEV1 vor Verabreichung des Studienmedikaments von Studienbeginn bis Tag 169 untersucht.
Eine integrierte Analyse der Daten beider Studien ergab für FF/VI 92/22 klinisch relevante Verbesserungen der Lungenfunktion. Zum Erhebungszeitpunkt an Tag 169 war unter FF/VI 92/22 sowie unter VI ein Anstieg des korrigierten mittleren Talspiegel-FEV1 um 129 ml (95%-KI: 91; 167 ml; p <0,001) bzw. 83 ml (95%-KI: 46; 121 ml; p <0,001) gegenüber Placebo feststellbar. FF/VI 92/22 bewirkte im Vergleich zu VI einen Anstieg des Talspiegel-FEV1 um 46 ml (95%-KI: 8; 83 ml; p= 0,017). An Tag 168 war unter FF/VI 92/22 sowie unter VI ein Anstieg des korrigierten mittleren gewichteten FEV1-Mittelwerts über 0 bis 4 Stunden um 193 ml (95%-KI: 156; 230 ml; p <0,001) bzw. 145 ml (95%-KI: 108; 181 ml; p <0,001) gegenüber Placebo feststellbar. FF/VI 92/22 bewirkte gegenüber FF alleine einen Anstieg des korrigierten mittleren gewichteten FEV1-Mittelwerts über 0 bis 4 Stunden um 148 ml (95%-KI: 112; 184 ml; p <0,001).
Zwölfmonatige Studien
HZC102970 und HZC102871 waren zwei 52-wöchige randomisierte, doppelblinde Parallelgruppenstudien zum Vergleich der Wirkungen von FF/VI 184/22, FF/VI 92/22 und FF/VI 46/22 mit VI 22, bei jeweils einmaliger täglicher Verabreichung, auf die Zahl der mittelschweren bis schweren Exazerbationen pro Jahr bei COPD-Patienten mit einer Raucheranamnese von mindestens 10 Packungsjahren und einem FEV1/FVC-Verhältnis nach Salbutamol von maximal 0,70 sowie einem FEV1 nach Salbutamol von höchstens 70% des Sollwerts und ≥1 anamnestisch dokumentierten COPD-Exazerbation in den 12 Monaten vor Visite 1, die mit Antibiotika und/oder oralen Kortikosteroiden behandelt werden musste oder eine Hospitalisierung erforderlich machte. Primärer Endpunkt war die Zahl der im Jahreszeitraum auftretenden mittelschweren und schweren Exazerbationen. Mittelschwere/schwere Exazerbationen waren definiert als Symptomverschlechterung, die eine Behandlung mit oralen Kortikosteroiden bzw. Antibiotika oder eine stationäre Aufnahme erforderlich machten. Beide Studien sahen eine 4-wöchige Vorbehandlungsphase («Run-in») vor, in der alle Teilnehmer unter offenen Bedingungen FP/Sal 250/50 zweimal täglich erhielten, um die COPD-Pharmakotherapie zu standardisieren und die Krankheit vor der Randomisierung für die verblindete 52-wöchige Studienbehandlung zu stabilisieren. Vor der Run-in-Phase setzten die Teilnehmer mit Ausnahme der kurzwirkenden Bronchodilatatoren alle Arzneimittel, die sie bislang aufgrund ihrer COPD erhalten hatten, ab. Die gleichzeitige Anwendung von langwirkenden inhalativen Bronchodilatatoren (Beta-2-Agonisten und Anticholinergika), Ipratropium/Salbutamol-Kombinationspräparaten, oralen Beta-2-Agonisten und Theophyllinpräparaten war während der Behandlungsphase nicht zulässig. Orale Kortikosteroide und Antibiotika waren unter Beachtung spezifischer Anwendungsleitlinien zur akuten Behandlung von COPD-Exazerbationen zulässig. Als Bedarfsmedikation verwendeten die Studienteilnehmer beider Studien während des gesamten Zeitraums Salbutamol.
Die Ergebnisse der beiden Studien zeigen, zusammengefasst in Tabelle 2, dass die Behandlung mit FF/VI 92/22 einmal täglich gegenüber VI zu einer niedrigeren jährlichen Rate an mittelschweren/schweren COPD Exazerbationen führte (p≤ 0,024).
Tabelle 2: Analyse der Exazerbationsraten nach 12-monatiger Behandlung
| Endpunkt | HZC102970 | HZC102871 | HZC102970 und HZC102871, integrierte Daten | |||||
|---|---|---|---|---|---|---|---|---|
| VI 22 (n= 409) | FF/VI 92/22 (n= 403) | VI 22 (n= 409) | FF/VI 92/22 (n= 403) | VI 22 (n= 818) | FF/VI 92/22 (n= 806) | |||
Mittelschwere und schwere Exazerbationen | ||||||||
| Korrigierte mittlere jährliche Rate | 1,14 | 0,90 | 1,05 | 0,70 | 1,11 | 0,81 | ||
| Verhältnis vs. VI | 0,79 | 0,66 | 0,73 | |||||
| 95%-KI | (0,64; 0,97) | (0,54; 0,81) | (0,63; 0,84) | |||||
| p-Wert | 0,024 | <0,001 | <0,001 | |||||
| % Reduktion | 21 | 34 | 27 | |||||
| 95%-KI | (3; 36) | (19; 46) | (16; 37) | |||||
Zeit bis zur ersten Exazerbation: | ||||||||
| Hazard-Ratio | 0,80 | 0,72 | 0,76 | |||||
| (95%-KI) | (0,66; 0,99) | (0,59; 0,89) | (0,66; 0,88) | |||||
| % Risikoreduktion | 20 | 28 | 24 | |||||
| p-Wert | 0,036 | 0,002 | p <0,001 | |||||
Exazerbationen mit Bedarf an systemischen/oralen Kortikosteroiden | ||||||||
| Jährliche Rate | 0,86 | 0,66 | 0,84 | 0,52 | 0,87 | 0,61 | ||
| Verhältnis vs. | 0,77 | 0,62 | 0,70 | |||||
| VI 95%-KI | (0,60; 0,99) | (0,49; 0,78) | (0,59; 0,83) | |||||
| p-Wert | 0,041 | <0,001 | <0,001 | |||||
| % Reduktion | 23 | 38 | 30 | |||||
| 95%-KI | (1; 40) | (22; 51) | (17; 41) | |||||
Im Rahmen dieser beiden Studien wurden auch Kombinationen von VI 22 mit FF 46 und FF 184 untersucht. Die Dosis-Wirkungs-Beziehung war nicht konsistent: Während eine Studie eine gewisse Dosisabhängigkeit mit dem besten Effekt bei der 184 µg Dosis zeigt, wurde in der anderen Studie der beste Effekt bei der 92 µg Dosis gemessen, während die 46 µg und die 184 µg Dosen ähnliche Effekte zeigten. Eine höhere Anzahl von Exazerbationen unter FF/VI 184/22 in der zweiten Studie HZC 102871 waren mit Pneumonien verbunden, welche teils fatalen verliefen. In der ersten Studie (HZC 102970) wurden in der FF/VI 184/22 Gruppe keine fatalen Pneumonien registriert.
Im Rahmen einer integrierten Analyse der Daten aus den Studien HZC102970 und HZC102871 wurde unter FF/VI 92/22 eine Verbesserung gegenüber VI 22 im Hinblick auf das korrigierte mittlere Talspiegel-FEV1 bei Woche 52 festgestellt (42 ml; 95%-KI: 0,019; 0,064; p <0,001). Die 24 Stunden anhaltende bronchodilatatorische Wirkung von FF/VI blieb von der ersten Gabe an über einen einjährigen Behandlungszeitraum ohne Anzeichen eines Wirksamkeitsverlusts (Tachyphylaxie) bestehen.
Vergleichsstudien mit Fluticasonpropionat/Salmeterol (FP/Sal)-Kombinationen
In einer 12-wöchigen Studie (HZC113107) an COPD-Patienten führten sowohl FF/VI 92/22 einmal täglich morgens als auch FP/Sal 500/50 µg zweimal täglich zu einer Verbesserung der Lungenfunktion gegenüber Studienbeginn. Die korrigierten mittleren Zunahmen des gewichteten FEV1-Mittelwerts über 0 bis 24 Stunden gegenüber Studienbeginn von 130 ml (FF/VI) bzw. 108 ml (FP/Sal) belegten eine allgemeine Verbesserung der Lungenfunktion über 24 Stunden unter beiden Behandlungen. Der korrigierte mittlere Behandlungsunterschied von 22 ml (95%-KI: -18; 63 ml) zwischen den Gruppen fiel statistisch nicht signifikant aus (p= 0,282).
Pharmakokinetik
Absorption
Die absolute Bioverfügbarkeit von Fluticasonfuroat (FF) und Vilanterol (VI) bei gleichzeitiger inhalativer Verabreichung als FF/VI betrug durchschnittlich 15,2% bzw. 27,3%. Die orale Bioverfügbarkeit von FF und VI war mit durchschnittlich 1,26% bzw. <2% gering. Angesichts dieser niedrigen oralen Bioverfügbarkeit ist die systemische Exposition gegenüber FF und VI nach Verabreichung per Inhalation hauptsächlich auf die Resorption des in die Lunge gelangten Anteils der verabreichten Dosis zurückzuführen.
Distribution
Nach intravenöser Verabreichung werden sowohl FF als auch VI umfassend verteilt, mit durchschnittlichen Verteilungsvolumina im Steady State von 661 L bzw. 165 L.
Sowohl FF als auch VI weisen eine niedrige Assoziation mit roten Blutkörperchen auf. In-vitro-Untersuchungen belegten mit durchschnittlich >99,6% bzw. 93,9% die hohe Plasmaproteinbindung von FF und VI in humanem Plasma. Bei Teilnehmern mit eingeschränkter Nieren- oder Leberfunktion war in vitro keine Abnahme der Plasmaproteinbindung feststellbar.
FF und VI sind Substrate des P-Glycoproteins (P-gp); allerdings ist aufgrund der guten Resorption beider Moleküle bei einer gleichzeitigen Verabreichung von FF/VI mit P-gp-Inhibitoren kaum mit einem Einfluss auf die systemische FF- bzw. VI-Exposition zu rechnen.
Metabolismus
Aus den In-vitro-Daten lässt sich ableiten, dass die Metabolisierung von FF und VI hauptsächlich über CYP3A4-vermittelte Reaktionswege erfolgt.
FF wird vorwiegend durch Hydrolyse der S-Fluoromethyl-carbothioat-Gruppe zu Metaboliten mit signifikant reduzierter Kortikosteroidwirkung abgebaut. VI wird in erster Linie durch O-Dealkylierung zu einer Reihe von Metaboliten abgebaut, die eine signifikant verminderte β1- und β2-agonistische Wirkung aufweisen.
Eine CYP3A4-Interaktionsstudie mit wiederholter Gabe wurde an gesunden Probanden mit FF/VI-184/22 und dem starken CYP3A4-Hemmer Ketoconazol (400 mg) durchgeführt. Durch die Ko‑Administration erhöhten sich die durchschnittliche AUC(0–24) und die durchschnittliche Cmax von FF um 36% bzw. 33%. Der Anstieg der FF-Exposition ging mit einer 27%igen Reduktion des gewichteten Serum-Cortisol-Mittelwerts von 0 bis 24 Stunden einher. Durch die Ko‑Administration erhöhten sich die durchschnittliche AUC(0-t) und die durchschnittliche Cmax von VI um 65% bzw. 22%. Der Anstieg der VI-Exposition war nicht mit einem Anstieg Beta-agonistisch bedingter systemischer Wirkungen auf Herzfrequenz, Kaliumkonzentrationen im Blut oder QTcF-Intervall assoziiert.
Elimination
FF wurde nach oraler Verabreichung beim Menschen hauptsächlich in Form seiner Metaboliten und fast ausschliesslich in den Fäzes ausgeschieden; <1% der radioaktiv markierten Dosis wurde im Urin wiedergefunden. Die scheinbare Plasma-Eliminationshalbwertszeit von FF nach inhalativer Verabreichung von FF/VI betrug durchschnittlich 24 Stunden.
VI wurde nach oraler Verabreichung hauptsächlich in Form seiner Metaboliten ausgeschieden; beim Menschen wurden ca. 70% einer radioaktiv markierten oralen Dosis im Urin und 30% im Stuhl wiedergefunden. Die scheinbare Plasma-Eliminationshalbwertszeit von VI nach inhalativer Verabreichung von FF/VI betrug durchschnittlich 2,5 Stunden. Die effektive Halbwertszeit für die Akkumulation von Vilanterol, bestimmt nach wiederholter Inhalation von 25 Mikrogramm-Dosen Vilanterol, beträgt bei Asthma-Patienten 16,0 Stunden und bei COPD-Patienten 21,3 Stunden.
Kinetik spezieller Patientengruppen
Kinder und Jugendliche
Für Jugendliche ab 12 Jahren ist keine Dosisänderung notwendig.
Die Pharmakokinetik, sowie Sicherheit und Wirksamkeit von Relvar Ellipta ist bei Patienten unter 12 Jahren nicht untersucht.
Ältere Patienten
Der Einfluss des Alters auf die Pharmakokinetik von FF und VI wurde in Phase-III-Studien zu COPD und Asthma untersucht. Es ergaben sich keine Hinweise auf einen altersbedingten (12–84 Jahre) Einfluss auf die Pharmakokinetik von Fluticasonfuroat und Vilanterol bei Asthma-Patienten.
Altersbedingte Auswirkungen auf die Pharmakokinetik von FF bei COPD-Patienten waren nicht feststellbar; dagegen kam es bei dem beobachteten Altersspektrum zwischen 41 und 84 Jahren zu einem Anstieg der AUC(0–24) von VI um 37%. Bei einem älteren Teilnehmer (84 Jahre) mit niedrigem Körpergewicht (35 kg) ist gegenüber dem Schätzwert für die untersuchte Population (COPD-Patient von 60 Jahren mit einem Gewicht von 70 kg) mit einer um 35% erhöhten VI-AUC(0–24) bei unveränderter Cmax zu rechnen. Diese Unterschiede sind wahrscheinlich nicht von klinischer Relevanz.
Für Asthma- und COPD-Patienten gibt es keine Empfehlungen bezüglich einer Dosisänderung.
Eingeschränkte Nierenfunktion
Aus einer Studie zur klinischen Pharmakologie von Relvar Ellipta geht hervor, dass eine hochgradige Nierenfunktionsbeeinträchtigung (Creatinin-Clearance <30 ml/min) gegenüber Nierengesunden nicht zu einer signifikant höheren Exposition gegenüber FF oder VI führt oder zu ausgeprägteren, durch Kortikosteroide oder Beta-2-Agonisten hervorgerufenen systemischen Wirkungen.
Bei Patienten mit beeinträchtigter Nierenfunktion ist keine Dosierungsanpassung erforderlich.
Die Auswirkungen der Hämodialyse wurden nicht untersucht.
Eingeschränkte Leberfunktion
Nach wiederholter Gabe von FF/VI über 7 Tage kam es bei Personen mit beeinträchtigter Leberfunktion (Child-Pugh-Stadium A, B oder C) gegenüber Lebergesunden zu einem Anstieg der systemischen FF-Exposition (bis zum Dreifachen, bezogen auf die AUC(0–24)). Der Anstieg der systemischen FF-Exposition unter FF/VI 184/22 bei Teilnehmern mit mässiger Leberfunktionsbeeinträchtigung (Child-Pugh-Stadium B) ging einher mit einer Reduktion der Cortisol-Serumkonzentrationen um durchschnittlich 34% im Vergleich zu lebergesunden Personen.
Bei Patienten mit schwerer Leberfunktionsbeeinträchtigung (Child-Pugh C), welche die niedrige Dosis von FF/VI 92/22 erhielten, zeigte sich keine Reduktion des Serum-Cortisolspiegels.
Bei Patienten mit mässiger oder schwerer Leberfunktionsbeeinträchtigung ist die Höchstdosis FF/VI 92/22 (siehe «Dosierung/Anwendung»).
Nach wiederholter Gabe von FF/VI über einen Zeitraum von 7 Tagen wurde bei Personen mit leichter, mässiger oder schwerer Leberfunktionseinschränkung (Child-Pugh-Stadium A, B oder C) kein signifikanter Anstieg der systemischen Exposition gegenüber VI (Cmax und AUC) beobachtet.
Im Vergleich zu Lebergesunden waren hinsichtlich der β-adrenergen systemischen Wirkungen (Herzfrequenz oder Serum-Kalium-Konzentrationen) keine klinisch relevanten Wirkungen der FF/VI-Kombination bei Patienten mit leichter bis mässiger (VI 22) oder schwerer Leberfunktionsbeeinträchtigung (VI 12,5) feststellbar.
Sonstige besondere Patientengruppen
Bei Asthma-Patienten lagen die Schätzwerte der Fluticasonfuroat-AUC(0–24) bei aus Ostasien, Japan und Südostasien stammenden Personen (12–13% der Studienteilnehmer) im Durchschnitt um 33% bis 53% höher als bei Personen anderer ethnischer Abstammung. Es gab allerdings keine Hinweise darauf, dass es im Zusammenhang mit der höheren systemischen Exposition bei dieser Population zu grösseren Auswirkungen auf die Cortisol-Ausscheidung im 24-Stunden-Urin kommt. Im Durchschnitt ist bei Personen asiatischer Abstammung gegenüber Personen anderer ethnischer Abstammung mit einer um 220 bis 287% erhöhten Vilanterol Cmax bei vergleichbarer AUC(0–24) zu rechnen. Es gab allerdings keine Hinweise darauf, dass diese höhere Cmax von Vilanterol zu klinisch relevanten Wirkungen auf die Herzfrequenz führt.
Bei COPD-Patienten lagen die Schätzwerte der FF-AUC(0–24) bei ostasiatischer, japanischer und südostasiatischer Abstammung (13–14% der Teilnehmer) im Durchschnitt um 23% bis 30% über denjenigen von Patienten weisser Hautfarbe. Es gab allerdings keine Hinweise darauf, dass es im Zusammenhang mit der höheren systemischen Exposition bei dieser Population zu grösseren Auswirkungen auf die Cortisol-Ausscheidung im 24-Stunden-Urin kommt. Bei COPD-Patienten war kein Einfluss der ethnischen Zugehörigkeit auf die Schätzwerte der pharmakokinetischen Parameter von VI feststellbar.
Geschlecht, Körpergewicht und BMI
In einer populationspharmakokinetischen Analyse der Daten aus Phase-III-Studien an 1'213 Asthma-Patienten (712 weiblich) und 1'225 COPD-Patienten (392 weiblich) ergaben sich keine Hinweise auf einen Einfluss von Geschlecht, Körpergewicht oder BMI (Body-Mass-Index) auf die Pharmakokinetik von FF.
In einer populationspharmakokinetischen Analyse der Daten von 856 Asthma-Patienten (500 weiblich) und 1'091 COPD-Patienten (340 weiblich) ergaben sich keine Hinweise auf einen Einfluss von Geschlecht, Körpergewicht oder BMI auf die Pharmakokinetik von VI.
Eine gewichts-, geschlechts-, oder BMI-abhängige Anpassung der Dosierung ist nicht erforderlich.
Präklinische Daten
Die pharmakologischen und toxikologischen Wirkungen, die in den präklinischen Studien unter Fluticasonfuroat (FF) oder Vilanterol (VI) beobachtet wurden, entsprechen den für Glukokortikoide bzw. Beta-2-Agonisten charakteristischen Wirkungen. Die kombinierte Verabreichung von FF mit VI führte nicht zu einer relevanten neuen toxischen Wirkung.
Karzinogenese, Mutagenese
FF hat sich bei der Ratte bzw. der Maus in einer Standard-Untersuchungsreihe als nicht genotoxisch und in Studien mit lebenslanger inhalativer Gabe bei Expositionen, die – bezogen auf die AUC – derjenigen des Menschen bei der empfohlenen Höchstdosis entsprechen, als nicht karzinogen erwiesen.
Aus Genotoxizitätsstudien geht hervor, dass VI keine genotoxischen Risiken für den Menschen birgt. Im Einklang mit den Ergebnissen für andere Beta-2-Agonisten wurden für VI in Studien mit lebenslanger inhalativer Gabe proliferative Wirkungen auf den Reproduktionstrakt weiblicher Ratten und Mäuse sowie auf die Hypophyse bei der Ratte beobachtet. Ein Anstieg der Tumorinzidenz wurde bei der Ratte bzw. der Maus bei Expositionen, die – bezogen auf die AUC – dem 1,2fachen bis 30fachen der menschlichen Exposition bei der empfohlenen Dosis entsprachen, nicht beobachtet.
Reproduktionstoxikologie
Die nach inhalativer Verabreichung von FF in Kombination mit VI an Ratten beobachteten Wirkungen entsprachen ungefähr denjenigen nach alleiniger Verabreichung von FF.
FF hat sich bei der Ratte bzw. beim Kaninchen als nicht teratogen erwiesen, führte aber bei maternotoxischer Dosierung bei Ratten zu einer Entwicklungsverzögerung und bei Kaninchen zum Abort. Wirkungen auf die Entwicklung von Ratten wurden bei Expositionen, die – bezogen auf die AUC – ungefähr dem Dreifachen der menschlichen Exposition bei der empfohlenen Höchstdosis entsprachen, nicht beobachtet
VI erwies sich bei der Ratte als nicht teratogen. In Inhalationsstudien an Kaninchen rief VI ähnliche Wirkungen hervor wie andere Beta-2-Agonisten (Gaumenspalten, offene Augenlider, Fusion der Sternebrae und verbogene Gliedmassen/Rotationsdeformitäten) bei Expositionen, die – bezogen auf die AUC – dem 14fachen der Exposition bei der empfohlenen Dosis im Menschen entsprachen. Bei subkutaner Gabe wurden bei Expositionen, die – bezogen auf die AUC – dem 84fachen der menschlichen Exposition bei der empfohlenen Dosis entsprachen, keine Wirkungen beobachtet.
Weder FF noch VI führten bei der Ratte zu unerwünschten Wirkungen auf die Fertilität oder auf die prä- und postnatale Entwicklung.
Die Ereignisse in juvenilen Tieren waren in der Regel typisch für ein Kortikosteroid oder einen Beta-2-Agonist, einschliesslich Effekten in stetig wachsenden oder sich entwickelnden Zähnen und die meisten Ereignisse wurden auch in Toxizitätsstudien mit erwachsenen Tieren beobachtet. Diese Erkenntnisse könnten darauf hindeuten, dass pädiatrische Patienten möglicherweise anfälliger für die Auswirkungen von inhalativen Kortikosteroiden sind und somit ein Anstieg der Zahnanomalien nicht ausgeschlossen werden kann.
Sonstige Hinweise
Haltbarkeit/besondere Lagerungshinweise
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Relvar Ellipta nicht über 25 °C und in der Originalverpackung aufbewahren, um den Inhalt vor Feuchtigkeit zu schützen. Die Schutzfolie darf erst unmittelbar vor dem ersten Gebrauch des Inhalators entfernt werden.
Haltbarkeit nach Öffnen des Schutzbehälters: 6 Wochen.
Das Datum, an dem der Schutzbehälter geöffnet worden ist, sollte auf der Etikette des Inhalators vermerkt werden, sobald dieser aus dem Schutzbehälter genommen wurde.
Arzneimittel für Kinder unzugänglich aufbewahren.
Hinweise für die Handhabung des Ellipta-Inhalators
Für eine ausführliche Anleitung zur Handhabung von Relvar Ellipta siehe Packungsbeilage.
Der Ellipta-Inhalator wird mit einem Trockenmittelpäckchen in einer Schale aus Verbundfolie dargereicht. In der Schale ist der Inhalator vor Feuchtigkeit geschützt; sie sollte daher erst unmittelbar vor der erstmaligen Anwendung geöffnet werden. Nach dem Öffnen muss das Trockenmittel-Päckchen entsorgt werden.
Die Schutzabdeckung des Ellipta-Inhalators darf nur geöffnet werden, wenn der Patient bereit ist zur Inhalation einer Dosis.
Wird die Schutzabdeckung des Ellipta-Inhalators geöffnet und geschlossen, ohne das Arzneimittel zu inhalieren, ist diese Dosis verloren. Die Dosis verbleibt zwar sicher im Inhalator, kann jedoch nicht mehr inhaliert werden. Die versehentliche Verabreichung einer zu hohen oder einer doppelten Dosis in einem Inhalationsvorgang ist dadurch ausgeschlossen.
Eine Überprüfung der korrekten Funktion oder eine besondere Vorbereitung des Ellipta-Inhalators vor der erstmaligen Anwendung sind nicht erforderlich.
Wichtiger Hinweis
Der Dosenzähler zeigt die Zahl der noch verbleibenden Dosen an. Wenn der Dosenzähler auf 05 steht, sollte ein neuer Inhalator besorgt werden. Wenn der Dosenzähler einen komplett rot ausgefüllten Hintergrund zeigt, muss der Inhalator gewechselt werden.
Zulassungsnummer
62969 (Swissmedic).
Zulassungsinhaberin
GlaxoSmithKline AG, 3053 Münchenbuchsee.
Stand der Information
Januar 2019.
Composition
Principes actifs: Fluticasoni-17 furoas, vilanterolum (ut Vilanteroli trifenatas).
Excipients: Lactosum monohydricum (avec de faibles quantités de protéines du lait), Magnesii stearas, q.s. ad pulverem.
Forme galénique et quantité de principe actif par unité
Poudre pour inhalation en récipient unidose.
Une dose unitaire de Relvar Ellipta 92/22 contient 100 µg de furoate de fluticasone et 25 µg de vilantérol (sous forme de trifénatate de vilantérol). La dose administrée (libérée par l'embout buccal de l'inhalateur Ellipta) est de 92 µg de furoate de fluticasone et de 22 µg de vilantérol.
Une dose unitaire de Relvar Ellipta 184/22 contient 200 µg de furoate de fluticasone et 25 µg de vilantérol (sous forme de trifénatate de vilantérol). La dose administrée (libérée par l'embout buccal de l'inhalateur Ellipta) est de 184 µg de furoate de fluticasone et de 22 µg de vilantérol.
Un inhalateur Ellipta contient 30 doses unitaires à inhaler.
Indications/Possibilités d’emploi
Asthme
Relvar Ellipta est utilisé dans le traitement régulier de l'asthme bronchique lorsqu'une association médicamenteuse (agoniste bêta-2 adrénergique à longue durée d'action et corticostéroïde inhalé) est indiquée: chez l'adulte et l'adolescent à partir d'un âge de 12 ans lorsqu'un traitement par des corticostéroïdes inhalés avec inhalation complémentaire d'un agoniste bêta-2 adrénergique à courte durée d'action comme médicament de secours ne suffit pas pour contrôler la maladie.
BPCO
Traitement symptomatique de la bronchopneumopathie chronique obstructive (BPCO) chez les patients présentant un VEMS <70% et ayant subi ≥2 exacerbations au cours des 12 derniers mois.
Posologie/Mode d’emploi
Relvar Ellipta est destiné exclusivement à l'inhalation (orale).
Relvar Ellipta est administré une fois par jour, toujours à la même heure de la journée.
Les patients doivent être instruits de se rincer la bouche à l'eau – sans avaler l'eau de rinçage – après l'inhalation. Ces rinçages permettent de prévenir le développement d'une candidose buccale et d'éviter les irritations de la gorge.
Asthme
Adultes et adolescents à partir de 12 ans:
Une inhalation de Relvar Ellipta 92/22 une fois par jour.
ou
Une inhalation de Relvar Ellipta 184/22 une fois par jour.
Une amélioration de la fonction pulmonaire est généralement constatable en l'espace de 15 minutes après l'inhalation de Relvar Ellipta. Les patients doivent toutefois être prévenus qu'un contrôle durable de leurs symptômes d'asthme exige une administration quotidienne régulière et que le traitement doit être poursuivi même si les symptômes ont disparu.
Si des symptômes de l'asthme apparaissent entre deux administrations du médicament, il convient d'utiliser un agoniste bêta-2-adrénergique à courte durée d'action pour atteindre un soulagement immédiat des symptômes.
Chez les adultes et les adolescents à partir de 12 ans qui ont besoin d'une dose faible à moyenne d'un corticostéroïde inhalé en association avec un agoniste bêta-2-adrénergique à longue durée d'action, Relvar Ellipta doit être prescrit à la dose initiale de 92/22 µg. Lorsqu'un patient n'atteint pas un contrôle suffisant sous Relvar Ellipta 92/22, on peut envisager une augmentation de la dose de 184/22 µg.
Les patients doivent continuer à être régulièrement évalués par le médecin afin de recevoir toujours le dosage optimal de furoate de fluticasone/vilantérol. La posologie ne doit être modifiée que sur prescription médicale. On choisira toujours la plus faible dose suffisante pour maintenir un contrôle efficace des symptômes. Le médecin prescripteur doit songer que dans le traitement de l'asthme, les effets d'une administration de furoate de fluticasone (FF) 92 µg une fois par jour sont comparables environ à ceux d'une administration de propionate de fluticasone (PF) 250 µg deux fois par jour et que FF 184 µg une fois par jour est comparable environ à PF 500 µg deux fois par jour.
Enfants de moins de 12 ans:
La sécurité et l'efficacité de Relvar Ellipta n'ont pas encore été suffisamment étudiées chez les enfants de moins de 12 ans.
BPCO
Adultes à partir de 40 ans:
Une inhalation de Relvar Ellipta 92/22 une fois par jour.
Les patients constatent généralement une amélioration de leur fonction pulmonaire en l'espace de 16 à 17 minutes après l'inhalation de Relvar Ellipta.
Relvar Ellipta 184/22 n'est pas homologué pour une utilisation dans le traitement de la BPCO. Aucun avantage supplémentaire de Relvar 184/22 par rapport à Relvar 92/22 n'a pu être démontré dans le traitement de la BPCO, tandis que les risques de pneumonie et d'effets indésirables systémiques liés au stéroïde pourraient être accrus (cf. «Mises en garde et précautions»).
Groupes de patients particuliers (asthme et BPCO)
Patients âgés (>65 ans):
Aucun ajustement de la posologie n'est nécessaire chez ce groupe de patients (cf. «Pharmacocinétique»).
Insuffisance rénale
Aucun ajustement de la posologie n'est nécessaire chez ce groupe de patients (cf. «Pharmacocinétique»).
Insuffisance hépatique
Une exposition systémique accrue au furoate de fluticasone (AUC) a été observée dans le cadre d'études chez des patients présentant une insuffisance hépatique légère, modérée ou sévère (cf. «Pharmacocinétique»).
La prudence est de rigueur lors d'une utilisation chez des patients insuffisants hépatiques, pour lesquels le risque d'effets indésirables systémiques liés au corticostéroïde peut éventuellement être accru. Chez les patients présentant une insuffisance hépatique modérée ou sévère, la dose maximale autorisée est d'une inhalation de Relvar Ellipta 92/22 une fois par jour.
Contre-indications
Relvar Ellipta est contre-indiqué chez les patients présentant une hypersensibilité connue à l'un des composants (cf. «Composition») ou une allergie sévère aux protéines du lait.
Mises en garde et précautions
Relvar Ellipta ne doit pas être utilisé pour le traitement des symptômes aigus de l'asthme ou des exacerbations aiguës de la BPCO. Ces situations exigent l'utilisation d'un bronchodilatateur à courte durée d'action. Lorsque l'utilisation d'un bronchodilatateur à courte durée d'action est de plus en plus souvent nécessaire pour soulager les symptômes, cela indique une réduction du contrôle des symptômes et exige une réévaluation des patients concernés par le médecin.
Les patients doivent être instruits de ne pas interrompre leur traitement par Relvar Ellipta sans surveillance médicale, étant donné que l'arrêt du médicament peut entraîner la réapparition des symptômes.
Des effets indésirables liés à l'asthme et des exacerbations de l'asthme peuvent se produire au cours du traitement par Relvar Ellipta. Les patients doivent être instruits de poursuivre le traitement, mais de consulter leur médecin si les symptômes d'asthme restent mal contrôlés ou s'aggravent après le début du traitement par Relvar Ellipta.
Bronchospasme paradoxal
Comme dans d'autres traitements par inhalation, un bronchospasme paradoxal avec aggravation immédiate des sibilances peut apparaître après l'administration. Ce phénomène doit être traité immédiatement par l'inhalation d'un bronchodilatateur à courte durée d''action. Dans un tel cas, le patient doit immédiatement interrompre le traitement par Relvar Ellipta, être soumis à un examen approfondi et recevoir au besoin un traitement alternatif.
Effets cardiovasculaires
Les médicaments sympathomimétiques tels que Relvar Ellipta peuvent provoquer des effets cardiovasculaires tels que des arythmies cardiaques (p.ex. tachycardie supraventriculaire et extrasystoles). De plus, on a rapporté sous agonistes bêta-2-adrénergiques comme Relvar Ellipta une survenue d'anomalies électrocardiographiques telles qu'un aplatissement de l'onde T, un allongement de l'intervalle QTc et un sous-décalage du segment ST. La signification clinique de ces anomalies reste cependant inconnue. Avant la prescription d'un traitement de fond par un agoniste bêta-2-adrénergique comme Relvar Ellipta, les patients atteints de BPCO devraient être examinés à la recherche d'éventuelles comorbidités cardiovasculaires. Par mesure de précaution, il est recommandé d'inclure à ces investigations un ECG avec recherche d'un éventuel allongement du QTc.
Patients présentant une insuffisance hépatique
Chez les patients présentant une insuffisance hépatique modérée ou sévère, on utilisera Relvar Ellipta 92/22 et surveillera l'apparition éventuelle d'effets indésirables systémiques liés au corticostéroïde (cf. «Posologie/Mode d'emploi»).
Effets systémiques des corticostéroïdes
L'administration simultanée de Relvar avec des inhibiteurs puissants du CYP3A4 doit être évitée.
Tous les corticostéroïdes inhalés peuvent provoquer des effets systémiques, surtout lors de la prescription au long cours d'une dose élevée. La probabilité d'une survenue de tels effets reste cependant plus faible qu'avec les corticostéroïdes oraux. Les effets systémiques possibles englobent une suppression de l'axe hypothalamo-hypophyso-surrénalien (HHS), une réduction de la densité minérale osseuse, ainsi qu'une cataracte et un glaucome, ou des maladies rares telles qu'une choriorétinopathie séreuse centrale (CRSC). Rarement, les corticostéroïdes inhalés peuvent provoquer des troubles psychiques, y compris une hyperactivité psychomotrice, des troubles du sommeil, une anxiété, une dépression ou un comportement agressif (en particulier chez les enfants).
Troubles visuels:
Des troubles visuels peuvent apparaître lors de l'utilisation systémique ou topique de corticostéroïdes. Si un patient se présente avec des symptômes tels qu'une vue floue ou d'autres troubles visuels, on envisagera de l'adresser à un ophtalmologue pour une évaluation des causes possibles; celles-ci incluent entre autres une cataracte, un glaucome ou des maladies rares telles qu'une CRSC qui ont été rapportées après l'utilisation de corticostéroïdes systémiques ou topiques.
Comme tous les médicaments contenant un corticoïde, Relvar Ellipta ne doit pas être administré sans les mesures de précaution qui s'imposent chez des patients atteints d'une tuberculose pulmonaire ou d'une infection chronique ou non traitée.
Hyperglycémie
Une glycémie accrue a été rapportée chez des patients diabétiques sous Relvar Ellipta. Cet aspect doit être pris en compte lors de la prescription de Relvar Ellipta à des patients diabétiques.
Pneumonies
Une incidence accrue de pneumonies a été observée sous Relvar Ellipta chez les patients atteints de BPCO. L'incidence des pneumonies exigeant une hospitalisation était également accrue. Ces pneumonies ont parfois eu une issue fatale (cf. «Effets indésirables»). Chez les patients atteints de BPCO, le médecin doit être attentif au développement possible d'une pneumonie, étant donné que les caractéristiques cliniques d'une telle infection sont partiellement les mêmes que les symptômes d'une exacerbation de la BPCO. Les facteurs de risque favorisant le développement d'une pneumonie chez les patients atteints de BPCO qui sont traités par Relvar Ellipta sont le tabagisme, un antécédent connu de pneumonie, un indice de masse corporelle <25 kg/m2 et un volume expiratoire maximal seconde (VEMS) <50% de la valeur prédite. Ces facteurs doivent être pris en compte lors de la prescription de Relvar Ellipta et la survenue d'une pneumonie doit conduire à une réévaluation du traitement.
Relvar Ellipta 184/22 n'est pas homologué pour une utilisation dans le traitement de la BPCO. Aucun avantage supplémentaire de Relvar Ellipta 184/22 par rapport à Relvar Ellipta 92/22 n'a pu être démontré dans le traitement de la BPCO, tandis que les risques de pneumonie et d'effets indésirables systémiques liés au corticostéroïde pourraient être accrus (cf. «Effets indésirables»).
La survenue de pneumonies a été fréquente chez les patients asthmatiques sous Relvar 184/22. Les pneumonies ont été numériquement plus nombreuses sous Relvar 184/22 que sous Relvar 92/22 ou sous placebo (cf. «Effets indésirables»). Aucun facteur de risque n'a pu être identifié.
Excipients
Relvar Ellipta contient du lactose. Les patients atteints d'une forme héréditaire rare d'intolérance au galactose, de déficit en lactase ou de malabsorption du glucose-galactose ne doivent pas utiliser Relvar Ellipta.
Interactions
Les études d'interactions n'ont été réalisées que chez des adultes.
Interactions pharmacocinétiques:
Bêtabloquants
Les bêtabloquants peuvent affaiblir ou antagoniser les effets des agonistes bêta-2-adrénergiques. L'utilisation concomitante d'un bêtabloquant non sélectif ou sélectif et de Relvar Ellipta doit être évitée, sauf si elle est absolument nécessaire.
Inhibiteurs du CYP3A4
Aussi bien le furoate de fluticasone que le vilantérol sont rapidement dégradés en large mesure par voie de l'enzyme hépatique CYP3A4 dans le cadre du métabolisme de premier passage.
L'administration simultanée avec des inhibiteurs puissants du CYP3A4 (p.ex. kétoconazole, itraconazole, clarithromycine, ritonavir ou médicaments contenant du cobicistat) peut accroître l'exposition aux corticostéroïdes et au vilantérol, et ainsi augmenter le risque d'effets indésirables, notamment ceux liés aux corticostéroïdes systémiques. L'administration simultanée doit être évitée à moins que le bénéfice attendu l'emporte sur le risque accru d'effets indésirables systémiques des corticostéroïdes; dans ce cas, les patients doivent être surveillés quant aux effets indésirables des corticostéroïdes systémiques (cf. «Pharmacocinétique»).
Inhibiteurs de la glycoprotéine P
Aussi bien le furoate de fluticasone que le vilantérol sont des substrats de la glycoprotéine P (P-gp). Une étude de pharmacologie clinique avec administration de vilantérol en association avec du vérapamil – un inhibiteur puissant de la P-gp et un inhibiteur modéré du CYP3A4 – chez des volontaires sains n'a révélé aucune influence importante sur la pharmacocinétique du vilantérol. Aucune étude de pharmacologie clinique n'a été effectuée avec un inhibiteur spécifique de la P-gp et le furoate de fluticasone.
Interactions pharmacodynamiques:
Médicaments connus pour provoquer un allongement de l'intervalle QTc
Comme pour d'autres agonistes bêta-2-adrénergiques, il existe théoriquement un risque d'interaction pharmacodynamique entre le vilantérol (contenu dans Relvar Ellipta) et les médicaments connus pour allonger l'intervalle QTC. Une telle interaction pourrait accroître le risque éventuel d'arythmies ventriculaires. Ces médicaments englobent par exemple certains antihistaminiques (p.ex. terfénadine, mizolastine), certains antiarythmiques (p.ex. quinidine), les phénothiazines, l'érythromycine, le ritonavir et les antidépresseurs tricycliques. L'administration supplémentaire de substances sympathomimétiques peut renforcer les effets indésirables cardiovasculaires. Si Relvar Ellipta est administré à des patients sous inhibiteurs de la MAO ou sous antidépresseurs tricycliques, la prudence est de rigueur parce que les effets des bêta-2-stimulants sur le système cardiovasculaire peuvent alors être renforcés.
L'administration en association avec la L-dopa, la L-thyroxine ou l'ocytocine peut avoir une influence négative sur la tolérance aux bêta-2-mimétiques.
Sympathomimétiques
L'utilisation d'autres sympathomimétiques (seuls ou comme composants d'un traitement d'association) pendant le traitement par Relvar Ellipta peut renforcer les effets indésirables du vilantérol.
Hypokaliémie
L'utilisation concomitante de Relvar Ellipta et d'un dérivé de méthylxanthine, d'un stéroïde ou d'un diurétique non épargneur de potassium pourrait renforcer une éventuelle hypokaliémie due aux agonistes bêta-2-adrénergiques.
Grossesse/Allaitement
Grossesse
On ne dispose que de données humaines limitées sur l'exposition pendant la grossesse.
Des expérimentations animales ont révélé une toxicité de reproduction après l'administration d'agonistes bêta-2-adrénergiques et de corticostéroïdes (cf. «Données précliniques»).
Relvar Ellipta ne doit pas être administré pendant la grossesse, sauf en cas de nécessité absolue.
Allaitement
Les données disponibles sur l'excrétion du furoate de fluticasone, du vilantérol et de leurs métabolites dans le lait maternel sont limitées. D'autres corticostéroïdes et agonistes bêta-2-adrénergiques sont toutefois détectables dans le lait maternel.
Un risque pour le nouveau-né/nourrisson allaité ne peut pas être exclu.
Il faut donc, après avoir soupesé les avantages de l'allaitement pour l'enfant et le bénéfice du traitement pour la mère, choisir d'arrêter soit l'allaitement, soit le traitement par Relvar Ellipta.
Effet sur l’aptitude à la conduite et l’utilisation de machines
Les effets de Relvar Ellipta sur l'aptitude à conduire des véhicules et à utiliser des machines n'ont pas été étudiés spécifiquement.
Effets indésirables
Les fréquences des effets indésirables en rapport avec Relvar Ellipta ont été déterminées à partir des données de grandes études cliniques sur l'asthme et la BPCO. Le programme de développement clinique pour le traitement de l'asthme a inclus les données d'un nombre total de 7034 patients dans une analyse intégrée des effets indésirables. Le programme de développement clinique pour le traitement de la BPCO a inclus les données d'un nombre total de 6237 patients dans une analyse intégrée des effets indésirables.
À l'exception des pneumonies et des fractures, le profil de sécurité a été similaire entre les patients asthmatiques et les patients atteints de BPCO. Dans les études cliniques, les pneumonies et les fractures ont été observées plus fréquemment chez les patients atteints de BPCO.
Les effets indésirables sont classés par systèmes d'organes et par fréquence. Les fréquences sont indiquées par ordre décroissant selon la convention suivante: très fréquents (>1/10), fréquents (≥1/100 à <1/10), occasionnels (≥1/1000 à <1/100), rares (≥1/10'000 à <1/1000), très rares (<1/10'000).
Au sein de chaque catégorie de fréquence, les effets indésirables sont indiqués par ordre décroissant de leur degré de sévérité.
Infections et infestations
Fréquents: pneumonie*, infections des voies respiratoires supérieures, bronchite, symptômes grippaux, candidose oropharyngée.
Affections du système nerveux
Très fréquents: céphalées (12 à 17%).
Affections cardiaques
Occasionnels: extrasystoles.
Affections respiratoires
Très fréquents: rhinopharyngite (10 à 14%).
Fréquents: douleurs oropharyngées, sinusite, pharyngite, rhinite, toux, enrouement.
Affections gastro-intestinales
Fréquents: douleurs abdominales.
Affections musculo-squelettiques
Fréquents: douleurs articulaires, douleurs dorsales, fractures**.
Troubles généraux et anomalies au site d'administration
Fréquents: fièvre.
Données post-commercialisation:
Affections du système immunitaire
Rares: réactions d'hypersensibilité, y compris anaphylaxie, angiœdème, rash et urticaire.
Troubles du métabolisme et de la nutrition
Occasionnels: hyperglycémie.
Affections psychiatriques
Fréquence indéterminée: anxiété.
Affections du système nerveux
Fréquence indéterminée: tremblements.
Affections cardiaques
Occasionnels: palpitations, tachycardie.
Affections musculo-squelettiques
Fréquents: crampes musculaires.
Affections respiratoires
Rares: bronchospasme paradoxal.
Description d'effets indésirables sélectionnés
* Pneumonie (cf. «Mises en garde et précautions»)
Dans deux études de 12 mois totalisant 3255 patients atteints de BPCO (VEMS moyen après l'administration d'un bronchodilatateur de 45% de la valeur prédite, écart type de 13%) qui avaient subi une exacerbation de la BPCO au cours de l'année précédente, l'incidence des pneumonies était plus élevée chez les patients traités par l'association de furoate de fluticasone (FF aux dosages de 46, 92 et 184 µg) et de vilantérol (VI) 22 µg que chez les patients traités uniquement par VI 22 µg (6% à 7% sous FF/VI versus 3% sous VI seul). Une pneumonie exigeant une hospitalisation a été observée chez 3% des patients sous FF/VI (tous les dosages) et chez <1% des patients sous VI seul. Neuf cas de pneumonie à issue fatale ont été rapportés dans le cadre de ces études. Sept de ces cas se sont produits sous FF/VI 184/22, un sous FF/VI 92/22 et un après la fin du traitement par VI seul.
Dans l'étude multicentrique, randomisée, SUMMIT (HZC113782) visant à évaluer la mortalité globale, 16 568 participants ont été traités par FF/VI 92/22, FF 92, VI 22 ou un placebo, en plus de bronchodilatateurs à la demande, pendant en moyenne 1,7 an. Les participants souffraient de BPCO modérée (VEMS moyen après l'administration d'un bronchodilatateur de 60% de la valeur prédite, ET 6%). Comme la plupart des patients avaient eu <2 exacerbations dans les 12 derniers mois (groupe de risque B selon la classification GOLD), il n'est que partiellement possible d'extrapoler les résultats concernant la survie et la sécurité à la population cible autorisée en Suisse (groupe de risque D selon la classification de GOLD).
Les effets indésirables de type pneumonie sont présentés dans le tableau suivant.
Evénements au cours du traitement | Nombre (%) de patients | |||
FF/VI 92/22 | FF 92 | VI 22 | Placebo | |
Pneumonie | 237 (6) [39,5] | 228 (5) [42,4] | 163 (4) [27,7] | 214 (5) [38,4] |
Pneumonie sévère | 140 (3) [22,4] | 146 (4) [25,1] | 104 (3) [16,4] | 127 (3) [22,2] |
Décès à la suite d'une pneumonie | 13 (<1) [1,8] | 10 (<1) [1,5] | 6 (<1) [0,9] | 9 (<1) [1,4] |
Dans une analyse intégrée des données de 11 études sur l'asthme (7034 patients), on a observé sous FF/VI 92/22 une incidence de pneumonies similaire à celle sous placebo (après correction en fonction de l'exposition, considérant les faibles nombres de cas et le nombre limité de patients sous placebo) (9,6/1000 années-patients sous FF/VI, 8,0/1000 années-patients sous placebo). L'incidence des pneumonies était plus élevée sous FF/VI 184/22 (18,4/1000 années-patients) que sous FF/VI 92/22 µg. Sous les deux doses, une hospitalisation n'a été nécessaire que dans peu de cas de pneumonie. Aucune différence concernant l'incidence d'événements sévères n'a été constatée entre les deux doses.
** Fractures
Dans deux études réitérées de 12 mois totalisant 3255 patients atteints de BPCO, l'incidence totale des fractures osseuses a été faible dans tous les groupes de traitement. Elle était plus élevée dans les groupes sous FF/VI (2%) que dans le groupe sous VI 22 µg (<1%). Bien que les groupes sous FF/VI aient présenté plus de fractures que le groupe sous VI 22 µg, les fractures typiquement associées aux corticostéroïdes (p.ex. fractures vertébrales comprimant la moelle épinière/thoraco-lombaires, fractures de la hanche et de l'acétabulum) ont été observées chez <1% des patients sous FF/VI et sous VI.
Les fractures observées dans l'étude SUMMIT (description, cf. ci-dessus) figurent dans le tableau suivant:
Evénements au cours du traitement | Nombre (%) de patients | |||
FF/VI 92/22 | FF 92 | VI 22 | Placebo | |
Toutes les fractures | 82 (2) [13,6] | 66 (2) [12,8] | 74 (2) [13,2] | 69 (2) [11,5] |
Fractures fréquemment associées à l'utilisation de CSI | 23 (<1) [3,4] | 24 (<1) [3,9] | 17 (<1) [2,4] | 13 (<1) [2,1] |
Dans une analyse intégrée des données de 11 études sur l'asthme (7034 patients), des fractures ont été observées avec une incidence <1% et étaient généralement associées à un traumatisme.
Surdosage
Signes et symptômes
On ne dispose pas de données d'études cliniques sur le surdosage de Relvar Ellipta.
Un surdosage de Relvar Ellipta peut causer des signes et symptômes correspondant aux effets de chacun des deux composants ou aux effets observés lors d'un surdosage d'autres agonistes bêta-2-adrénergiques ainsi qu'aux. effets de classe connus des corticostéroïdes inhalés (cf. «Mises en garde et précautions»).
Traitement
Aucun traitement spécifique n'est disponible pour le cas d'un surdosage de Relvar Ellipta. Lors d'un surdosage, le patient doit recevoir le traitement de soutien approprié à son cas et être surveillé en conséquence.
Un traitement bêtabloquant cardiosélectif ne doit être envisagé que lors d'un surdosage sévère de vilantérol, causant des symptômes cliniques sérieux et ne répondant pas aux mesures de soutien. Chez les patients ayant des antécédents connus de bronchospasmes, la prudence est de rigueur lors de l'utilisation d'un bêtabloquant cardiosélectif.
La marche à suivre ultérieure dépendra des exigences cliniques ou, le cas échéant, des recommandations du centre d'information toxicologique.
Propriétés/Effets
Code ATC: R03AK10
Mécanisme d'action
Le furoate de fluticasone et le vilantérol sont des représentants de deux classes de médicaments différentes (l'un étant un corticostéroïde synthétique et l'autre un agoniste des récepteurs bêta-2-adrénergiques sélectif à longue durée d'action).
Effets pharmacodynamiques
Furoate de fluticasone (FF)
Le FF est un corticostéroïde synthétique trifluoré aux effets anti-inflammatoires puissants. On ignore quel est exactement le mécanisme par lequel le FF influence les symptômes de l'asthme et de la BPCO. Il est démontré que les corticostéroïdes ont un large spectre d'action influençant différents types de cellules (p.ex. éosinophiles, macrophages, lymphocytes) et différents médiateurs (p.ex. les cytokines et chimiokines impliquées dans les processus inflammatoires).
Trifénatate de vilantérol (VI)
Le VI est un agoniste bêta-2-adrénergique sélectif à longue durée d'action (LABA).
Les effets pharmacologiques des agonistes des récepteurs bêta-2-adrénergiques tels que le VI reposent du moins partiellement sur la stimulation de l'adénylate cyclase intracellulaire, une enzyme catalysant la transformation de l'adénosine triphosphate (ATP) en adénosine-3′,5′-monophosphate cyclique (AMPc). Les concentrations accrues d'AMPc provoquent une relaxation de la musculature lisse bronchique et inhibent la libération (en particulier par les mastocytes) de médiateurs impliqués dans les réactions d'hypersensibilité immédiate.
Efficacité clinique
Asthme
Trois études randomisées de phase III (HZA106827, HZA106829 et HZA106837), réalisées en double aveugle et de durées différentes, ont examiné la sécurité et l'efficacité de Relvar Ellipta (furoate de fluticasone/vilantérol, FF/VI) chez des patients adultes et adolescents souffrant d'asthme persistant. Tous les participants avaient utilisé un CSI (corticostéroïde inhalé) avec ou sans LABA pendant au moins 12 semaines avant la visite 1. Tous les participants de l'étude HZA106837 avaient subi au moins une exacerbation ayant exigé un traitement par des corticostéroïdes oraux au cours de l'année précédant la visite 1. L'étude HZA106827 était une étude de 12 semaines pour évaluer l'efficacité de l'association FF/VI 92/22 µg (n = 201) et du FF 92 seul (n = 205) versus placebo (n = 203) dans le cadre d'une administration une fois par jour. L'étude HZA106829 était une étude de 24 semaines pour évaluer l'efficacité de l'association FF/VI 184/22 µg (n = 197) et du FF 184 seul (n = 194) administrés une fois par jour versus propionate de fluticasone 500 µg (PF 500) administré deux fois par jour (n = 195).
Les études HZA106827/HZA106829 ont examiné chez tous les participants, en tant que critères co-primaires, les variations du VEMS au moment des concentrations sanguines minimales (peu avant l'administration du bronchodilatateur ou de la médication de l'étude) dans le cadre des visites à la clinique du début de l'étude jusqu'à la fin de la phase de traitement, ainsi que, chez un sous-groupe de participants, la valeur moyenne pondérée de la série de mesures du VEMS de 0 à 24 heures après l'administration à la fin de la phase de traitement. Les variations (en %) des tranches de 24 h sans utilisation d'une médication de secours pendant la phase de traitement versus valeurs initiales étaient un critère secondaire ayant permis une discrimination statistique suffisante. Les résultats des critères primaires et des critères secondaires les plus importants de ces études sont résumés dans le tableau 1.
Tableau 1: Résultats des critères primaires et des critères secondaires les plus importants des études HZA106827 et HZA106829
| N° de l'étude | HZA106829 | HZA106827 | ||
|---|---|---|---|---|
| Dose du traitement par FF/VI* (µg) | FF/VI 184/22 une fois par jour vs FF 184 une fois par jour | FF/VI 184/22 une fois par jour vs PF 500 deux fois par jour | FF/VI 92/22 une fois par jour vs FF 92 une fois par jour | FF/VI 92/22 une fois par jour vs placebo une fois par jour |
Variations du VEMS au moment de la concentration sanguine minimale versus valeurs initiales, approche LOCF (report de la dernière valeur obtenue) | ||||
| Différence entre les traitements | 193 ml | 210 ml | 36 ml | 172 ml |
| Valeur p | p <0,001 | p <0,001 | p= 0,405 | p <0,001 |
| (IC à 95%) | (108; 277) | (127; 294) | (-48; 120) | (87; 258) |
Valeur moyenne pondérée de la série de mesures du VEMS de 0 à 24 h après l'administration | ||||
| Différence entre les traitements | 136 ml | 206 ml | 116 ml | 302 ml |
| Valeur p | p= 0,048 | p= 0,003 | p= 0,06 | p <0,001 |
| (IC à 95%) | (1; 270) | (73; 339) | (-5; 236) | (178; 426) |
Variations (en %) des tranches de 24 h sans utilisation d'une médication de secours versus valeurs initiales | ||||
| Différence entre les traitements | 11,7% | 6,3% | 10,6% | 19,3% |
| Valeur p | p <0,001 | p= 0,067 | p <0,001 | p <0,001 |
| (IC à 95%) | (4,9; 18,4) | (-0,4; 13,1) | (4,3; 16,8) | (13,0; 25,6) |
Variations (en %) des tranches de 24 h sans symptômes versus valeurs initiales | ||||
| Différence entre les traitements | 8,4% | 4,9% | 12,1% | 18,0% |
| Valeur p | p= 0,010 | p= 0,137 | p <0,001 | p <0,001 |
| (IC à 95%) | (2,0; 14,8) | (-1,6; 11,3) | (6,2; 18,1) | (12,0; 23,9) |
Variations du VEMS (volume expiratoire maximal seconde) matinal versus valeurs initiales | ||||
| Différence entre les traitements | 33,5 l/min | 32,9 l/min | 14,6 l/min | 33,3 l/min |
| Valeur p | p <0,001 | p <0,001 | p <0,001 | p <0,001 |
| (IC à 95%) | (25,3; 41,7) | (24,8; 41,1) | (7,9; 21,3) | (26,5; 40,0) |
Variations du VEMS (volume expiratoire maximal seconde) en soirée versus valeurs initiales | ||||
| Différence entre les traitements | 30,7 l/min | 26,2 l/min | 12,3 l/min | 28,2 l/min |
| Valeur p | p <0,001 | p <0,001 | p <0,001 | p <0,001 |
| (IC à 95%) | (22,5; 38,9) | (18,0; 34,3) | (5,8; 18,8) | (21,7; 34,8) |
* FF/VI = furoate de fluticasone/vilantérol = Relvar Ellipta
Dans l'étude HZA106837, la durée de traitement était variable (au moins 24 semaines à au maximum 76 semaines; la majorité des patients ont reçu au moins 52 semaines de traitement). Les patients de l'étude HZA106837 ont été randomisés pour recevoir soit l'association FF/VI 92/22 (n = 1009), soit du FF 92 (n = 1010) une fois par jour. Le critère primaire de l'étude HZA106837 était le temps écoulé jusqu'à la première exacerbation sévère de l'asthme. Une exacerbation sévère de l'asthme était définie comme une aggravation des symptômes d'asthme ayant exigé une utilisation de corticostéroïdes systémiques pendant au moins trois jours, une hospitalisation ou une visite aux urgences pour un traitement systémique de l'asthme. Les variations moyennes corrigées du VEMS au moment de la concentration sanguine minimale versus valeurs initiales ont également été analysées en tant que critère secondaire.
Dans l'étude HZA106837, le risque d'une exacerbation sévère de l'asthme était réduit de 20% sous FF/VI 92/22 versus FF 92 seul (hazard ratio de 0,795; p = 0,036; IC à 95%: 0,642; 0,985). La fréquence des exacerbations sévères de l'asthme par patient et par année était de 0,19 (environ 1 cas tous les 5 ans) sous FF 92 seul et de 0,14 (environ 1 cas tous les 7 ans) sous FF/VI 92/22. Le rapport des taux d'exacerbation entre le FF/VI 92/22 et le FF 92 était de 0,755 (IC à 95%: 0,603; 0,945). La fréquence des exacerbations sévères de l'asthme était donc réduite de 25% sous FF/VI 92/22 versus FF 92 (p = 0,014). Les effets bronchodilatateurs de Relvar Ellipta – persistants sur 24 heures – sont restés identiques, sans signes d'une perte d'efficacité (tachyphylaxie) jusqu'à la fin de la période de traitement d'un an. En comparaison avec le FF 92 seul, on a observé sous FF/VI 92/22 une amélioration homogène de 83 ml à 95 ml des valeurs du VEMS au moment de la concentration sanguine minimale dans les semaines 12, 36 et 52 ainsi qu'à la fin de l'observation (p <0,001; IC à 95%: 52; 126 ml à la fin de l'observation). À la fin du traitement, 40% des patients sous FF/VI 92/22 présentaient un bon contrôle des symptômes (ACQ7 ≤0,75), par rapport à 36% des patients sous FF 92 (p <0,001; IC à 95%: 1,23; 1,82).
Études comparatives avec le propionate de fluticasone/salmétérol (PF/Sal)
Dans une étude de 24 semaines (HZA113091) auprès de patients adultes et adolescents souffrant d'asthme persistant, une amélioration de la fonction pulmonaire versus valeurs initiales a été atteinte aussi bien sous FF/VI 92/22 une fois par jour (en soirée) que sous PF/Sal 250/50 µg deux fois par jour. Les augmentations moyennes corrigées de la moyenne pondérée du VEMS de 0 à 24 heures versus valeurs initiales, de 341 ml (FF/VI 92/22) et de 377 ml (PF/Sal 250/50), ont reflété une amélioration globale de la fonction pulmonaire sur 24 heures sous chacun des deux traitements. La différence moyenne corrigée entre les traitements, de 37 ml, n'était pas statistiquement significative (p = 0,162). Pour la valeur du VEMS au moment de la concentration sanguine minimale, les patients du groupe sous FF/VI ont atteint une variation moyenne de 281 ml versus valeur initiale et les patients du groupe sous PF/Sal ont atteint une variation de 300 ml (la différence de la valeur moyenne ajustée entre les groupes, de 19 ml (IC à 95%‑: -0,073; 0,034), n'était pas statistiquement significative (p = 0,485)).
Aucune étude comparative adéquate versus PF/Sal ou versus d'autres associations de CSI/LABA n'a été effectuée pour comparer les effets sur les exacerbations de l'asthme.
Furoate de fluticasone en monothérapie
Une étude de 24 semaines (FFA112059), randomisée, en double aveugle et contrôlée contre placebo, a examiné la sécurité et l'efficacité du FF 92 administré une fois par jour (n = 114) et du PF 250 administré deux fois par jour (n = 114) versus placebo (n = 115) chez des patients adultes et adolescents souffrant d'asthme persistant. Tous les participants devaient avoir été traités depuis au moins 4 semaines par un CSI à dose fixe lors de la visite 1 (examen préliminaire); l'utilisation d'un LABA était interdite dans les 4 semaines précédant la visite 1. Le critère primaire d'efficacité était défini comme la variation du VEMS mesuré dans le cadre des visites cliniques au moment de la concentration sanguine minimale (avant l'administration du bronchodilatateur et de la dose) entre le début de l'étude et la fin de la phase de traitement. Les variations (en %) des tranches de 24 h sans utilisation d'une médication de secours pendant la phase de traitement de 24 semaines versus valeurs initiales étaient un critère secondaire ayant permis une discrimination statistique suffisante. Lors de l'évaluation au bout de 24 semaines, le VEMS mesuré au moment de la concentration sanguine minimale était augmenté respectivement de 146 ml sous FF (IC à 95%: 36; 257 ml; p = 0,009) et de 145 ml sous PF (IC à 95%: 33; 257 ml; p = 0,011) par rapport au placebo. Aussi bien le FF que le PF ont permis une augmentation du pourcentage de tranches de 24 h sans utilisation d'une médication de secours. Les augmentations étaient respectivement de 14,8% (IC à 95%: 6,9; 22,7; p <0,001) et de 17,9% (IC à 95%: 10,0; 25,7; p <0,001) par rapport au placebo.
Étude de provocation par un allergène
Les effets bronchoprotecteurs de l'association FF/VI 92/22 contre les réactions asthmatiques de type immédiat et tardif à un allergène inhalé ont été examinés dans une étude croisée contrôlée contre placebo, avec administration répétée de 4 variantes de traitement (HZA113126) chez des patients souffrant d'un asthme léger. Les patients ont été randomisés pour recevoir une fois par jour pendant 21 jours un traitement par FF/VI 92/22, par FF 92, par VI 22 ou par placebo. Un test de provocation par un allergène a été effectué une heure après l'administration de la dernière dose. Comme allergènes, on a utilisé – selon les résultats individuels au test – des acariens de la poussière de maison, des squames de chat ou du pollen de bouleau. Les valeurs des séries de mesure du VEMS ont été comparées à celles obtenues après l'inhalation d'une simple solution saline avant la provocation par l'allergène. Les plus grands effets sur la réaction asthmatique de type immédiat ont été observés sous FF/VI 92/22 versus FF 92 seul et versus VI 22 seul. Aussi bien l'association FF/VI 92/22 que le FF 92 seul ont pratiquement empêché la réaction asthmatique tardive versus vilantérol seul. Lors du test de provocation à la métacholine, effectué le jour 22, l'association FF/VI 92/22 a exercé une protection significativement plus puissante contre l'hyperréactivité bronchique induite par l'allergène que les monothérapies par FF ou par VI.
BPCO
Le programme de développement clinique pour le traitement de la BPCO a inclus une étude de 12 semaines (HZC113107), deux études de 6 mois (HZC112206, HZC112207) et deux études d'un an randomisées et contrôlées (HZC102970, HZC102871) auprès de patients atteints d'une BPCO diagnostiquée cliniquement. Ces études ont examiné entre autres la fonction pulmonaire, la dyspnée et le taux d'exacerbations modérées à sévères.
Études de six mois
Les études HZC112206 et HZC112207 étaient des études de 24 semaines, randomisées, en double aveugle, contrôlées versus placebo, en groupes parallèles, pour examiner les effets de l'association FF/VI versus chacune des monosubstances et versus placebo. Dans HZC112206, l'efficacité des associations FF/VI 46/22 (n = 206) et FF/VI 92/22 (n = 206) a été comparée avec celle du FF 92 (n = 206), du VI 22 (n = 205) et d'un placebo (n = 207), toujours dans le cadre d'une administration une fois par jour. Dans HZC112207, l'efficacité des associations FF/VI 92/22 (n = 204) et FF/VI 184/22 (n = 205) a été comparée avec celle du FF 92 (n = 204), du FF 184 (n = 203), du VI 22 (n = 203) et du placebo (n = 205), toujours dans le cadre d'une administration une fois par jour.
Tous les patients devaient avoir au moment de l'examen préliminaire des antécédents de tabagisme d'au moins 10 paquets-années, un rapport VEMS/CVF maximal de 0,70 après administration de salbutamol, un VEMS maximal de 70% de la valeur prédite après administration de salbutamol et un score de dyspnée ≥2 à l'échelle MRC modifiée (modified Medical Research Council, mMRC; échelle de 0 à 4). Lors de l'examen préliminaire pour les études HZC112206 et HZC112207, le VEMS moyen avant l'administration d'un bronchodilatateur était de 42,6% et de 43,6% de la valeur prédite et la réversibilité moyenne était de 15,9% et de 12,0% respectivement. Les deux études ont examiné en tant que critères co-primaires la valeur moyenne pondérée du VEMS de 0 à 4 heures après l'administration le jour 168 ainsi que les variations de la valeur du VEMS au moment du taux sanguin minimal (avant l'administration du médicament de l'étude) entre le début de l'étude et le jour 169.
Une analyse intégrée des données des deux études a montré des améliorations cliniquement significatives de la fonction pulmonaire sous FF/VI 92/22. Lors de l'évaluation effectuée le jour 169, on a constaté une augmentation du VEMS moyen corrigé au moment de la concentration sanguine minimale chez les patients sous FF/VI 92/22 et sous VI seul. Cette augmentation était de 129 ml (IC à 95%: 91; 167 ml; p <0,001) sous FF/VI 92/22 et de 83 ml (IC à 95%: 46; 121 ml; p <0,001) sous VI seul versus placebo. En comparaison avec le VI, l'association FF/VI 92/22 a permis une augmentation de 46 ml du VEMS au moment de la concentration sanguine minimale (IC à 95%: 8; 83 ml; p = 0,017). Le jour 168, on a constaté une augmentation de la valeur moyenne pondérée du VEMS de 0 à 4 heures de 193 ml (IC à 95%: 156; 230 ml; p <0,001) sous FF/VI 92/22 et de 145 ml (IC à 95%: 108; 181 ml; p <0,001) sous VI seul versus placebo. En comparaison avec le FF seul, le FF/VI 92/22 a permis une augmentation de 148 ml de la valeur moyenne pondérée du VEMS de 0 à 4 heures (IC à 95%: 112; 184 ml; p <0,001).
Études de douze mois
Les études HZC102970 et HZC102871 étaient deux études de 52 semaines, randomisées, en double aveugle, en groupes parallèles, pour examiner les effets des associations FF/VI 184/22, FF/VI 92/22 et FF/VI 46/22 versus VI 22, toujours dans le cadre d'une administration une fois par jour, sur le nombre annuel d'exacerbations modérées à sévères chez des patients atteints de BPCO qui avaient des antécédents de tabagisme d'au moins 10 paquets-années, un rapport VEMS/CVF maximal de 0,70 après administration de salbutamol, un VEMS maximal de 70% de la valeur prédite après administration de salbutamol et ≥1 antécédent documenté d'exacerbation de la BPCO – ayant exigé l'administration d'antibiotiques et/ou de corticostéroïdes oraux ou une hospitalisation – dans les 12 mois ayant précédé la visite 1. Le critère primaire était le nombre d'exacerbations modérées ou sévères survenues en l'espace d'un an. Les exacerbations modérées/sévères étaient définies comme une aggravation des symptômes exigeant un traitement par des corticostéroïdes oraux et/ou des antibiotiques ou une hospitalisation. Les deux études avaient prévu une phase préalable de 4 semaines (phase de «run-in») pendant laquelle tous les participants ont été traités en ouvert par PF/Sal 250/50 deux fois par jour pour obtenir une standardisation de la pharmacothérapie contre la BPCO et stabiliser la maladie avant la randomisation pour le traitement en aveugle de 52 semaines de l'étude. Avant la phase de run-in, les participants ont cessé l'administration de tout médicament utilisé jusque-là pour le traitement de la BPCO à l'exception des bronchodilatateurs à courte durée d'action. L'administration concomitante de bronchodilatateurs inhalés à longue durée d'action (agonistes bêta-2-adrénergiques ou anticholinergiques), d'associations d'ipratropium/salbutamol, d'agonistes bêta-2-adrénergiques oraux ou de théophylline était interdite pendant la phase de traitement. Les corticostéroïdes oraux et les antibiotiques étaient autorisés pour le traitement aigu des exacerbations de la BPCO en respectant des directives d'utilisation spécifiques. En tant que médication de secours, les participants aux deux études ont utilisé du salbutamol pendant toute la durée de leur participation.
Les résultats de ces deux études, résumés dans le tableau 2, montrent que le traitement par FF/VI 92/22 administré une fois par jour a permis d'atteindre un plus faible taux annuel d'exacerbations modérées/sévères de la BPCO que le traitement par VI (p ≤0,024).
Tableau 2: Analyse des taux d'exacerbations au bout de 12 mois de traitement
| Critère | HZC102970 | HZC102871 | HZC102970 et HZC102871, données intégrées | |||
| VI 22 (n= 409) | FF/VI 92/22 (n= 403) | VI 22 (n= 409) | FF/VI 92/22 (n= 403) | VI 22 (n= 818) | FF/VI 92/22 (n= 806) | |
Exacerbations modérées et sévères | ||||||
| Taux annuel moyen corrigé | 1,14 | 0,90 | 1,05 | 0,70 | 1,11 | 0,81 |
| Rapport vs VI | 0,79 | 0,66 | 0,73 | |||
| IC à 95% | (0,64; 0,97) | (0,54; 0,81) | (0,63; 0,84) | |||
| Valeur p | 0,024 | <0,001 | <0,001 | |||
| % de réduction | 21 | 34 | 27 | |||
| IC à 95% | (3; 36) | (19; 46) | (16; 37) | |||
Temps écoulé jusqu'à la première exacerbation | ||||||
| Hazard ratio | 0,80 | 0,72 | 0,76 | |||
| (IC à 95%) | (0,66; 0,99) | (0,59; 0,89) | (0,66; 0,88) | |||
| % de réduction du risque | 20 | 28 | 24 | |||
| Valeur p | 0,036 | 0,002 | p <0,001 | |||
Exacerbations exigeant des corticostéroïdes systémiques/oraux | ||||||
| Taux annuel | 0,86 | 0,66 | 0,84 | 0,52 | 0,87 | 0,61 |
| Rapport vs VI | 0,77 | 0,62 | 0,70 | |||
| IC à 95% | (0,60; 0,99) | (0,49; 0,78) | (0,59; 0,83) | |||
| Valeur p | 0,041 | <0,001 | <0,001 | |||
| % de réduction | 23 | 38 | 30 | |||
| IC à 95% | (1; 40) | (22; 51) | (17; 41) | |||
Les associations FF/VI 46/22 et FF/VI 184/22 ont également été examinées dans le cadre de ces deux études. Le rapport dose-effet n'a pas été cohérent: alors qu'une des deux études suggère une certaine dépendance de la dose avec le meilleur effet à la dose de 184 µg, l'autre a mesuré le meilleur effet à la dose de 92 µg, tandis que les doses de 46 µg et de 184 µg ont eu des effets similaires. Dans la deuxième étude (HZC 102871), un nombre plus élevé d'exacerbations sous FF/VI 184/22 était associé à des pneumonies, dont certaines ont eu une issue fatale. Aucune pneumonie à issue fatale n'a été observée sous FF/VI 184/22 dans la première étude (HZC 102970).
Une analyse intégrée des données des études HZC102970 et HZC102871 a montré une amélioration de la valeur moyenne corrigée du VEMS au moment du taux sanguin minimal à la semaine 52 sous FF/VI 92/22 versus VI 22 (42 ml; IC à 95%: 0,019; 0,064; p <0,001). Les effets bronchodilatateurs de l'association FF/VI – persistants sur 24 heures – sont restés identiques, sans signes d'une perte d'efficacité (tachyphylaxie) de la première administration jusqu'à la fin de la période de traitement d'un an.
Études comparatives avec les associations de propionate de fluticasone/salmétérol (PF/Sal)
Dans une étude de 12 semaines (HZC113107) auprès de patients atteints de BPCO, une amélioration de la fonction pulmonaire versus valeurs initiales a été atteinte aussi bien sous FF/VI 92/22 une fois par jour (le matin) que sous PF/Sal 500/50 µg deux fois par jour. Les augmentations moyennes corrigées de la moyenne pondérée du VEMS de 0 à 24 heures versus valeurs initiales, de 130 ml (FF/VI) et de 108 ml (PF/Sal), ont reflété une amélioration globale de la fonction pulmonaire sur 24 heures sous chacun des deux traitements. La différence moyenne corrigée de 22 ml entre les deux traitements (IC à 95%: -18; 63 ml) n'était pas statistiquement significative (p = 0,282).
Pharmacocinétique
Absorption
La biodisponibilité absolue du furoate de fluticasone (FF) et du vilantérol (VI) administrés simultanément par inhalation de l'association FF/VI était en moyenne de 15,2% et de 27,3% respectivement. La biodisponibilité orale du FF et du VI était faible, en moyenne de 1,26% et de <2% respectivement. En raison de cette faible biodisponibilité orale, l'exposition systémique au FF et au VI après leur administration par inhalation est due essentiellement à la résorption de la partie de la dose administrée parvenue dans les poumons.
Distribution
Après administration intraveineuse, aussi bien le FF que le VI sont distribués largement, avec des volumes de distribution moyens de 661 l et de 165 l respectivement à l'état d'équilibre.
Le FF et le VI ont présenté tous deux une faible association aux globules rouges. Des essais in vitro ont démontré une forte liaison aux protéines plasmatiques, atteignant en moyenne >99,6% pour le FF et 93,9% pour le VI dans le plasma humain. Aucune réduction de la liaison aux protéines plasmatiques n'a été constatée in vitro chez les participants présentant une insuffisance rénale ou hépatique.
Le FF et le VI sont des substrats de la glycoprotéine P (P-gp), mais la bonne résorption des deux molécules ne fait guère supposer qu'une administration concomitante de FF/VI et d'inhibiteurs de la P-gp influence l'exposition systémique au FF et au VI.
Métabolisme
Les données in vitro permettent de conclure que la métabolisation du FF et du VI s'effectue essentiellement par des voies réactionnelles dépendant du CYP3A4.
Le FF est surtout dégradé par hydrolyse de la fonction S-fluorométhyl-carbothioate et transformé ainsi en métabolites présentant des effets corticostéroïdes significativement réduits. Le VI est surtout dégradé par O-désalkylation en une série de métabolites présentant une activité agoniste β1- et β2-adrénergique significativement réduite.
Une étude auprès de volontaires sains a été effectuée pour examiner les interactions au niveau du CYP3A4 entre l'association FF/VI-184 /22 et le kétoconazole (400 mg) – un inhibiteur puissant du CYP3A4 – lors d'administrations répétées. La co-administration a conduit à une augmentation de 36% de l'AUC(0-24) moyenne et de 33% de la Cmax moyenne du FF. L'augmentation de l'exposition au FF a été associée à une réduction de 27% de la valeur moyenne pondérée de cortisol sérique de 0 à 24 heures. La co-administration a conduit à une augmentation de 65% de l'AUC(0-t) moyenne et de 22% de la Cmax moyenne du VI. L'augmentation de l'exposition au VI n'a pas été associée à une augmentation des effets agonistes bêta-adrénergiques systémiques sur la fréquence cardiaque, les concentrations sanguines de potassium ou l'intervalle QTcF.
Élimination
Le FF administré par voie orale chez l'être humain a été éliminé essentiellement sous forme de métabolites, presque exclusivement dans les selles; <1% de la dose radiomarquée a été retrouvée dans les urines. La demi-vie d'élimination plasmatique apparente du FF administré par inhalation de FF/VI a été de 24 heures en moyenne.
Le VI administré par voie orale a été éliminé essentiellement sous forme de métabolites; chez l'être humain, environ 70% d'une dose orale radiomarquée ont été retrouvés dans les urines et 30% dans les selles. La demi-vie d'élimination plasmatique apparente du VI administré par inhalation de FF/VI a été de 2,5 heures en moyenne. La demi-vie effective d'accumulation du vilantérol, déterminée à la suite d'inhalations répétées de doses de 25 µg de vilantérol, est de 16,0 heures chez les patients asthmatiques et de 21,3 heures chez les patients atteints de BPCO.
Cinétique pour certains groupes de patients
Enfants et adolescents
Aucun ajustement posologique n'est nécessaire chez les adolescents à partir d'un âge de 12 ans.
La pharmacocinétique, la sécurité et l'efficacité de Relvar Ellipta n'ont pas été étudiées chez les enfants de moins de 12 ans.
Patients âgés
L'influence de l'âge sur la pharmacocinétique du FF et du VI a été étudiée dans des études de phase III auprès de patients souffrant d'asthme ou de BPCO. Il n'en est ressorti aucun indice suggérant une influence de l'âge (de 12 à 84 ans) sur la pharmacocinétique du furoate de fluticasone et du vilantérol chez les patients asthmatiques.
Chez les patients atteints de BPCO, aucune influence de l'âge n'a été constatée sur la pharmacocinétique du FF, mais une augmentation de 37% de l'AUC(0-24) du VI a été observée dans la fourchette d'âges étudiée de 41 à 84 ans. Chez un patient âgé (84 ans) et présentant un faible poids corporel (35 kg), il faut s'attendre à une augmentation de 35% de l'AUC(0-24) du VI en comparaison avec la valeur estimée chez la population étudiée (patients avec BPCO, âgés de 60 ans et pesant 70 kg), tandis que la Cmax reste inchangée. Ces différences ne sont sans doute pas cliniquement significatives.
Il n'existe pas de recommandations posologiques concernant une modification de la dose chez les patients asthmatiques ou atteints de BPCO.
Insuffisance rénale
Une étude sur la pharmacologie clinique de Relvar Ellipta suggère qu'une insuffisance rénale sévère (clairance de la créatinine <30 ml/min) n'entraîne pas une exposition significativement accrue au FF ou au VI en comparaison avec les personnes sans insuffisance rénale et qu'elle ne provoque pas d'effets systémiques plus marqués du corticostéroïde ou de l'agoniste bêta-2-adrénergique.
Aucun ajustement de la dose n'est nécessaire chez les patients insuffisants rénaux.
Les effets de l'hémodialyse n'ont pas été étudiés.
Insuffisance hépatique
Après l'administration répétée de FF/VI pendant 7 jours, les personnes avec une insuffisance hépatique (stades Child-Pugh A, B ou C) ont présenté une exposition systémique accrue au FF, pouvant aller d'après l'AUC(0-24) jusqu'au triple de l'exposition observée chez les personnes sans insuffisance hépatique. Chez les participants présentant une insuffisance hépatique modérée (stade Child-Pugh B), l'augmentation de l'exposition systémique au FF sous FF/VI 184/22 était associée à une réduction de 34% en moyenne des concentrations sériques de cortisol par rapport aux personnes sans insuffisance hépatique. Aucune réduction du taux sérique de cortisol n'a été constatée sous la faible dose de FF/VI 92/22 chez les patients présentant une insuffisance hépatique sévère (Child-Pugh C).
Chez les patients présentant une insuffisance hépatique modérée ou sévère, la dose maximale est de FF/VI 92/22 (cf. «Posologie/Mode d'emploi»).
Après l'administration répétée de FF/VI pendant 7 jours, aucune augmentation significative de l'exposition systémique au VI (Cmax et AUC) n'a été observée chez les personnes présentant une insuffisance hépatique légère, modérée ou sévère (stades Child-Pugh A, B ou C).
En comparaison avec les personnes sans insuffisance hépatique, on n'a pas constaté d'effets bêta-adrénergiques systémiques (fréquence cardiaque ou taux sériques de potassium) cliniquement significatifs sous l'association FF/VI chez les patients présentant une insuffisance hépatique légère à modérée (VI 22) ou sévère (VI 12,5).
Autres groupes particuliers de patients
Dans le traitement de l'asthme, les valeurs estimées de l'AUC(0-24) du furoate de fluticasone chez les personnes originaires d'Asie de l'Est, du Japon et d'Asie du Sud-est (12 à 13% des participants aux études) étaient en moyenne supérieures de 33% à 53% à celles chez les personnes d'autres origines ethniques. Aucun indice n'a toutefois suggéré que les expositions systémiques supérieures chez cette population soient associées à une influence importante sur l'élimination de cortisol dans les urines de 24 heures. On doit s'attendre en moyenne à une augmentation de 220 à 287% de la Cmax du vilantérol, en présence d'une AUC(0-24) comparable, chez les personnes d'origine asiatique versus autres origines ethniques. Aucun indice n'a toutefois suggéré que cette Cmax accrue du vilantérol entraîne des effets cliniquement importants sur la fréquence cardiaque.
Parmi les patients atteints de BPCO, les personnes originaires d'Asie de l'Est, du Japon et de l'Asie du Sud-est (13 à 14% des participants) avaient des valeurs estimées de l'AUC(0-24) du FF qui étaient en moyenne supérieures de 23% à 30% à celles des Blancs. Aucun indice n'a toutefois suggéré que les expositions systémiques supérieures chez cette population soient associées à une influence importante sur l'élimination de cortisol dans les urines de 24 heures. Chez les patients atteints de BPCO, aucune influence de l'appartenance ethnique sur les valeurs estimées des paramètres pharmacocinétiques du VI n'a été constatable.
Sexe, poids corporel et IMC
Dans une analyse de données pharmacocinétiques de population provenant d'études de phase III auprès de 1213 patients asthmatiques (dont 712 de sexe féminin) et 1225 patients atteints de BPCO (dont 392 de sexe féminin), aucun indice n'a suggéré une influence du sexe, du poids corporel ou de l'IMC (indice de masse corporelle) sur la pharmacocinétique du FF.
Dans une analyse de données pharmacocinétiques de population de 856 patients asthmatiques (dont 500 de sexe féminin) et de 1091 patients atteints de BPCO (dont 340 de sexe féminin), aucun indice n'a suggéré une influence du sexe, du poids corporel ou de l'IMC sur la pharmacocinétique du VI.
Un ajustement posologique en fonction du poids, du sexe ou de l'IMC n'est pas nécessaire.
Données précliniques
Les effets pharmacologiques et toxicologiques observés dans les études précliniques sous furoate de fluticasone (FF) ou sous vilantérol (VI) correspondent aux effets caractéristiques des glucocorticoïdes et des agonistes bêta-2-adrénergiques. L'administration de FF en association avec le VI n'a provoqué aucun nouvel effet toxique important.
Carcinogénicité, mutagénicité
Le FF n'a montré aucune génotoxicité dans une série d'études standard sur des rats et des souris et n'a montré aucune carcinogénicité dans des études avec inhalation pendant la vie entière avec des expositions correspondant – d'après l'AUC – à la dose maximale recommandée chez l'homme.
Il ressort des études de génotoxicité que le VI ne présente pas de risques génotoxiques pour l'être humain. En accord avec les résultats obtenus pour d'autres agonistes bêta-2-adrénergiques, les études avec administration de VI par inhalation pendant la vie entière ont constaté des effets prolifératifs sur l'appareil reproducteur chez les rats et les souris femelles ainsi que sur l'hypophyse chez les rats. Aucune augmentation de l'incidence de cancers n'a été observée chez les rats et les souris lors d'expositions correspondant respectivement – d'après l'AUC – à 1,2 fois et à 30 fois l'exposition humaine à la dose recommandée.
Toxicologie de reproduction
Après l'administration par inhalation de FF/VI chez des rats, les effets observés étaient environ les mêmes qu'après l'administration de FF seul.
Le FF n'a pas été tératogène chez le rat et le lapin. Aux doses toxiques pour la mère, il a cependant provoqué des retards du développement chez le rat et des avortements chez le lapin. Aucun effet sur le développement des rats n'a été observé à des expositions correspondant environ – d'après l'AUC – au triple de l'exposition humaine à la dose maximale recommandée.
Le VI n'a pas été tératogène chez le rat. Dans des études d'inhalation chez le lapin, le VI a provoqué des effets similaires à ceux d'autres agonistes bêta-2-adrénergiques (fentes palatines, paupières ouvertes, fusion des sternèbres et déformation par rotation/courbure de membres) à des expositions correspondant – d'après l'AUC – à 14 fois l'exposition humaine à la dose recommandée. Aucun effet n'a été observé lors d'une administration sous-cutanée à des expositions correspondant – d'après l'AUC – à 84 fois l'exposition humaine à la dose recommandée.
Ni le FF ni le VI n'ont provoqué chez le rat des effets indésirables sur la fertilité ou le développement prénatal et postnatal.
Les événements observés chez les animaux juvéniles étaient généralement typiques d'un corticostéroïde ou d'un agoniste bêta-2-adrénergique (y compris effets sur les dents en cours de développement ou de croissance continue) et la plupart des événements ont aussi été observés dans les études de toxicité chez les animaux adultes. Ces observations suggèrent que les patients pédiatriques pourraient éventuellement être plus sensibles aux effets des corticostéroïdes inhalés et qu'une augmentation des anomalies dentaires ne peut donc pas être exclue.
Remarques particulières
Conservation/Remarques concernant le stockage
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Ne pas conserver Relvar Ellipta au-dessus de 25 °C et le conserver dans son emballage d'origine pour le protéger de l'humidité. Le film protecteur ne doit être retiré que juste avant la première utilisation de l'inhalateur.
Conservation après ouverture de la barquette de protection: 6 semaines
La date d'ouverture de la barquette de protection doit être inscrite sur l'étiquette de l'inhalateur, dès que celui-ci a été retiré de la barquette de protection.
Conserver le médicament hors de la portée des enfants.
Remarques concernant la manipulation de l'inhalateur Ellipta
Voir la notice d'emballage pour un mode d'emploi détaillé de Relvar Ellipta.
L'inhalateur Ellipta est fourni avec un sachet dessiccant dans une barquette en feuille composite. La barquette protège l'inhalateur de l'humidité et ne doit donc être ouverte que juste avant la première utilisation. Une fois que l'emballage a été ouvert, le sachet dessiccant doit être jeté.
Le couvercle de sécurité de l'inhalateur Ellipta ne doit être ouvert que lorsque le patient est prêt à inhaler une dose.
Si le couvercle de sécurité de l'inhalateur Ellipta est ouvert, puis refermé sans que le médicament soit inhalé, la dose sera perdue. Cette dose restera enfermée dans l'inhalateur en toute sécurité, mais ne pourra plus être inhalée. Ainsi, l'administration accidentelle d'une dose trop élevée ou d'une double dose en une seule inhalation est exclue.
Il n'est pas nécessaire de procéder à une vérification du fonctionnement correct ou à une préparation particulière de l'inhalateur Ellipta avant la première utilisation.
Remarque importante
Le compteur des doses indique le nombre de doses encore contenues dans le dispositif. Lorsque le compteur de doses indique 05, il faut se procurer un nouvel inhalateur. Lorsque le compteur de doses présente un fond entièrement rouge, il faut passer au nouvel inhalateur.
Numéro d’autorisation
62969 (Swissmedic).
Titulaire de l’autorisation
GlaxoSmithKline AG, 3053 Münchenbuchsee.
Mise à jour de l’information
Janvier 2019.
Reviews (0)

Free consultation with an experienced pharmacist
Describe the symptoms or the right drug - we will help you choose its dosage or analogue, place an order with home delivery or just consult.
We are 14 pharmacists and 0 bots. We will always be in touch with you and will be able to communicate at any time.
Bestsellers

Milupa Aptamil Pepti Syneo Can 400g
Product code: 7748334Aptamil Pepti Syneo is a special food for babies from birth who are allergic to cow's milk protein a..
55.08 CHF


Weleda Baby Calendula Wind- und Wetterbalsam 30ml
Product code: 5455461Der Weleda Baby Calendula Wind- und Wetterbalsam schützt die sensible Babyhaut intensiv bei Kälte un..
15.35 CHF


HerpoTherm herpes pen
Product code: 7798882Herpotherm® - the heating pen Cold sores are not only unattractive but can also be very painful. Th..
78.48 CHF

Burgerstein Vitamin D3 capsules 600 IE 100 pieces
Product code: 6091601Because the sun doesn't shine every day. Burgerstein D3 600 IU are capsules and contain vitamin D in..
26.51 CHF



